 许娇娇,黄百芬,周健,项瑜芝,任一平,蔡增轩.直接稀释-超高效液相色谱-串联质谱法快速测定谷物及其制品中16种真菌毒素[J].中国食品卫生杂志,2017,29(6):708-715.
许娇娇,黄百芬,周健,项瑜芝,任一平,蔡增轩.直接稀释-超高效液相色谱-串联质谱法快速测定谷物及其制品中16种真菌毒素[J].中国食品卫生杂志,2017,29(6):708-715.DOi:10.13590/j.cjfh.2017.06.015
直接稀释-超高效液相色谱-串联质谱法快速测定谷物及其制品 中16种真菌毒素
(1.浙江省疾病预防控制中心,浙江 杭州310051; 2.宁波市疾病预防控制中心, 浙江 宁波315010; 3.浙江工业大学化学工程学院,浙江 杭州310014; 4.浙江清华长三角 研究院 国家食品安全风险评估中心应用技术合作中心,浙江 嘉兴314006)
收稿日期:2017-09-12
作者简介:许娇娇女主管技师研究方向为食品检验 E-mail:jjxucdc@163.com
通信作者:蔡增轩男副研究员研究方向为食品卫生理化检验 E-mail:caizx_cdc@163.com
基金项目:国家重大科学仪器设备开发专项项目(2011YQ060084)
摘要:目的建立谷物及其制品中黄曲霉毒素、赭曲霉毒素、单端孢霉烯族类毒素、伏马菌素等16种真菌毒素的超高效液相色谱-串联质谱(UPLC-MS/MS)检测方法。方法5 g样品加入20 ml乙腈-水-甲酸(79∶20∶1,V/V)溶液提取,10 000 r/min离心5 min后稀释,Cortecs C18色谱柱(100 mm×3.0 mm,1.6 μm)分离,以乙腈-0.1%甲酸水作为流动相梯度洗脱,采用四极杆串联质谱仪电喷雾模式检测,同位素稀释内标法定量。结果16种真菌毒素在线性范围内(0.05~200 ng/ml)具有良好的线性关系,相关系数(r2)>0.99,方法定量限为0.1~50 μg/kg,加标回收率为72.67%~126.92%之间,相对标准偏差(RSD)为0.15%~16.83%。应用本方法分析英国弗帕斯检测技术研究所(FAPAS)质控样品,测定结果均在标示范围内,方法准确度良好。结论本方法具有良好的灵敏度、回收率和重复性,前处理方法简单,适用于常规实验室对谷物及其制品中真菌毒素的检测,满足日常真菌毒素监测工作的需要。
关键词:
谷物及其制品; 真菌毒素; 超高效液相色谱-串联质谱法; 直接稀释; 食品污染物; 测定
文章编号:1004-8456(2017)06-0708-08
中图分类号:R155
文献标志码:A
A dilute-and shoot approach using ultra high-performance liquid chromatograph-mass/mass spectrometry for 16 mycotoxins analysis in cereals and products
(1.Zhejiang Provincial Center of Disease Control and Prevention,Zhejiang Hangzhou 310051,China; 2.Ningbo Municipal Center for Disease Control and Prevention, Zhejiang Ningbo 315010, China; 3.Zhejiang University of Technology, Zhejiang Hangzho)
Abstract:ObjectiveTo develop and validate the ultra high-performance liquid chromatograph coupled with mass spectrometry (UPLC-MS/MS) method for simultaneous determination of 16 mycotoxins, including aflatoxins, ochratoxins, trichothecenes, fumonisins and so on. MethodsFive grams of the sample was extracted with acetonitrile/water/formic acid (79∶ 20∶ 1, V/V). After centrifuged and diluted, the extraction was separated by Cortecs C18(100 mm×3.0 mm,1.6 μm) analytical column under acetonitrile-0.1% formic acid gradient eluting, then was determined by UPLC-MS/MS. Dilution isotope internal calibration was used for qualification. ResultsThe result showed good linearities (0.05-200 ng/ml) for 16 mycotoxins in the certain correlation ranges with the coefficients all above 0.99. The limits of quantification ranged from 0.1 to 50 μg/kg. The average recoveries of 16 mycotoxins at three spiked levels ranged in 72.67%-126.92% with relative standard deviations of 0.15%-16.83%. The accuracies were acceptable by detecting natural matrix quality control samples. ConclusionThe method is rapid, simple, sensitive, reproducible and accurate for simultaneous determination of multiple mycotoxins in cereals and products in routine laboratories. It meets the the requirement of mycotoxins monitoring in relative food.
Key words:
Cereals and products; mycotoxins; ultra high performance liquid chromatography-tandem mass spectrometry; dilute-and shoot; food contaminants; determination
真菌毒素是一类由产毒丝状真菌在适宜的环境条件下产生的次级代谢产物,目前已知的化合物有300多种。根据联合国粮农组织估计,全世界每年约有25%的粮食作物受到真菌毒素的污染,造成达数千亿美元的经济损失,同时也对人体和动物的健康具有极大的直接或潜在危害,因此,世界各国相继规定了常见的十几种真菌毒素的限量[1]。其中,欧盟已制定了包括黄曲霉毒素(aflatoxins,AFTs)、单端孢霉烯族类[如脱氧雪腐镰刀菌烯醇(deoxynivalenol, DON)、T-2毒素和HT-2毒素]、伏马菌素(fumonisins, FBs)、赭曲霉毒素A(ochratoxin A, OTA)以及玉米赤霉烯酮(zearalenone, ZEN)等毒素在加工和未加工粮食作物及其制品中的最大限量[2]。国际食品法典委员会(CAC)制定了包括DON、T-2、HT-2、ZEN、AFTs以及FBs在内的一些常见真菌毒素的最大限量以及每日耐受摄入量[3-4]。我国GB 2761—2017《食品安全国家标准 食品中真菌毒素限量》[5]中也规定了食品中DON(1 000 μg/kg)、ZEN(60 μg/kg)、AFTB1(20 μg/kg)以及OTA(5 μg/kg)的最大限量。
真菌毒素的检测技术从早期的仅针对单一毒素检测的酶联免疫(ELISA)法、薄层色谱法、荧光光度法[6],到可检测同一类毒素多个化合物的气相或液相色谱法[7-8],再到目前被广泛应用于多组分化合物同时定量分析的液相色谱-串联质谱(LC-MS/MS)法[9-11],方法的灵敏度、准确性以及便捷性都有了较大的提高和发展。基于液相色谱-串联质谱的检测方法是准确定量检测真菌毒素的主要手段,其优势在于该方法具有灵敏度高、定量准确和抗干扰能力强等特点。传统的样品前处理净化策略(例如固相萃取柱净化和免疫亲和柱净化)往往仅适用于一种或特定类型的毒素化合物[12]。已有文献[13-16]报道采用直接稀释-液相色谱-串联质谱法同时检测食品中多种真菌毒素化合物,这些方法往往需要超过20 min的色谱分析时间,或仅覆盖十种甚至更少的毒素种类。本试验旨在建立一种适用于多种食品基质的多真菌毒素快速检测方法,以更短的色谱分析时间,定量分析涵盖我国现行真菌毒素限量标准[5](GB 2761—2017)以及欧盟法规[2]中涉及的真菌毒素及其相关衍生物,从而更高效、全面地提供我国食品中真菌毒素污染情况信息。
本试验全面优化前处理以及色谱、质谱各环节技术参数,建立适用于常规实验室应用的同位素内标定量检测粮谷类食品中16种真菌毒素的方法,可为我国食品中多真菌毒素风险监测与暴露评估提供技术支持。
AFTB1(CAS:1162-65-8)、AFTB2(CAS:7220-81-7)、AFTG1(CAS:1165-39-5)、AFTG2(CAS:7241-98-7)、雪腐镰刀菌烯醇(NIV,CAS:023282-20-4)、DON(CAS:51481-10-8)及其乙酰化衍生物(3-ADON,CAS:50722-38-8;15-ADON,CAS:88337-96-6)、ZEN(CAS:017924-92-408)、OTA(CAS:000303-47-9)、FB1(CAS:116355-83-0)、FB2(CAS:116355-84-1)、FB3(CAS:136379-59-4)、T-2毒素(CAS:021259-20-1)、HT-2毒素(CAS:026934-87-2)和杂色曲霉素(ST,CAS:10048-13-2)16种真菌毒素标准品以及相应的15种13C同位素内标标准溶液(纯度≥98%,无15-ADON的同位素内标标准溶液)均购自奥地利RomeLab,乙腈、甲醇、甲酸均为色谱纯,试验用水为超纯水,玉米粉参考物质[T2298QC和T4230QC,英国弗帕斯检测技术研究所(FAPAS)]。
混合标准储备液:取各单一标准储备液2 ml于10 ml容量瓶,用乙腈定容至刻度,即得混合标准储备液。
混合同位素内标工作液:分别移取一定体积的同位素标准溶液,用乙腈稀释定容,得到混合同位素内部工作液,其中13C-AFTB1、13C-AFTB2、13C-AFTG1、13C-AFTG2、13C-OTA含量为0.01 μg/ml,13C-NIV、13C-DON、 13C-3ADON、13C-ZEN含量为1.25 μg/ml,13C-ST含量为0.02 μg/ml,13C-T-2含量为0.02 μg/ml,13C-HT-2、 13C-FB1、13C-FB2、13C-FB3含量为0.5 μg/ml。
标准曲线溶液:准确移取混合标准储备液适量,采用20%乙腈-水溶液逐级稀释,配制成分别含AFTB1 0.05、0.1、0.2、0.5、1.0、2.0 ng/ml的标准曲线系列溶液。移取20 μl同位素内标混合溶液于内插管中,加入180 μl对应的混合标准曲线浓度点溶液,混匀即得标准曲线溶液系列。
质谱:电喷雾离子源(ESI),毛细管电压2.5 kV(ESI+)/3.5 kV(ESI-),离子源温度为150 ℃,脱溶剂气温度为500 ℃,脱溶剂气流速为800 L/h,多反应监测模式(MRM)。
大部分化合物以[M+H]+作为母离子,而T-2毒素以[M+NH4]+离子响应丰度较高。HT-2毒素既存在[M+H]+离子模式,也存在[M+NH4]+离子模式,经考察比较,这2种模式根据质谱设备状态的不同而产生不同的响应丰度。ZEN在正负离子模式均有较好的响应,但13C-ZEN在正离子模式下产生337>301子离子,该子离子通道对ZEN子离子318>301通道有干扰,而在负离子模式下该同位素化合物对ZEN子离子通道无干扰,因此,ZEN化合物最终采用[M-H]-负离子模式。
流动相的差异会直接影响到目标化合物的离子化效率。根据文献[9,13,15-16]报道以及各化合物的分子结构特征,乙酸铵或甲酸铵的加入可以抑制加钠峰[M+Na]+的形成,酸的加入有利于改善如FBs这一类酸性毒素的峰型;因此,本研究考察了甲酸溶液(0.1%)、乙酸铵溶液(5 mmol/L)、甲酸(0.1%)-乙酸铵溶液(5 mmol/L),以及乙腈、甲醇不同有机溶剂对各化合物的离子化效率的影响。如图1所示,采用甲醇作为有机相时,大部分单端孢霉烯族类毒素(如DON、3-ADON、15-ADON以及HT-2)响应灵敏度较高,而其他的毒素,特别是AFTs、OTA这两类限量值较低的毒素,在乙腈作为流动相时,响应灵敏度较甲醇条件下的高。甲醇的洗脱能力弱于乙腈,当用甲醇作为强洗脱溶剂时,整个色谱分离时间会相对较长。从图1中还可以看到,5 mmol/L的乙酸铵仅可以增强T-2和ST毒素的响应,但0.1%甲酸作为水相溶剂时,其他大部分毒素都有较好的响应。为了保证目标分析物的响应最佳和缩短分析时间,本研究最终选择0.1%甲酸水-乙腈溶液为流动相。
对于多真菌毒素的液相色谱-串联质谱检测分析,色谱柱的选择依然重要。虽然质谱方法可以根据其质量参数的专一性对某些色谱上难以分离的化合物进行区分,但对于3-ADON和15-ADON这一类同分异构体,母离子质量数完全相同,子离子相似,从而通道间有干扰,因此,为了能够进行准确定量分析,仍需要从色谱行为上加以区分。本试验分别考察了Waters ACQUITY UPLC BEH C18(3.0 mm×100 mm,1.7 μm)、 Waters ACQUITY UPLC HSS T3(3.0 mm×100 mm,1.7 μm)和Waters Cortecs UPLC C18(3.0 mm×100 mm,1.6 μm)3种色谱柱对16种毒素的分离效果。结果显示选择Cortecs C18能够使得3-ADON和15-ADON达到基线分离,并在15 min内完成16种真菌毒素的色谱质谱分离定量。该色谱条件既达到同分异构体基线分离的目的,也减少了试剂用量,环境友好,还提高了工作效率。
甲醇和乙腈水溶液体系是真菌毒素提取过程中最常用的有机溶剂,但实际试验中显示乙腈对蛋白沉淀效果以及提取液离心后的澄清度均优于甲醇,故本试验对不同比例的乙腈-水溶液进行了考察。图2结果表明,对大多数毒素化合物,提取溶液有机相比例在70%~90%之间,均有较好的回收率。亲水性较强的如呕吐类毒素,提取溶液水相比例高时,提取回收率最佳;而亲脂性较强的如ZEN化合物,提取溶液有机相比例高时,提取回收率最佳。综合考虑16种真菌毒素的最佳平均提取回收率,故本试验采用80%乙腈-水溶液作为提取溶液。
多真菌毒素的前处理条件,除了考虑提取溶液的有机相比例,还需考虑提取液的酸度。真菌毒素中的FBs、OTA等化合物含有羧基,在酸性条件下更容易以分子形式进入有机相。另外,有研究[7]表明呕吐类毒素以及T-2毒素在碱性条件下可能会发生形态转变,故而提取溶液中加入适量的酸将有助于提高提取效率和化合物的稳定。本研究考察了不同酸性条件下提取溶液对于16种真菌毒素提取效率的影响,结果如图3所示。提取液中甲酸的比例对16种毒素的提取效率有着不同的影响:呕吐类毒素的提取效率无明显影响;随着提取液中甲酸浓度的增加,T-2毒素的回收率明显降低;其他的大部分毒素在一定范围的甲酸浓度下,提取回收率随着甲酸浓度的增加而提高,但当甲酸浓度高于5%时,提取效率降低。因此,1%甲酸提取液为本试验中最佳的提取溶液。
本试验考察了10、20、30和40 min不同提取时间对提取效率的影响。本试验采用涡旋振荡器提取多种毒素,结果显示,提取时间为30 min时达到 最高提取回收率,提取时间为40 min时回收率没有明显差异。
真菌毒素的检测技术从早期的仅针对单一毒素检测的酶联免疫(ELISA)法、薄层色谱法、荧光光度法[6],到可检测同一类毒素多个化合物的气相或液相色谱法[7-8],再到目前被广泛应用于多组分化合物同时定量分析的液相色谱-串联质谱(LC-MS/MS)法[9-11],方法的灵敏度、准确性以及便捷性都有了较大的提高和发展。基于液相色谱-串联质谱的检测方法是准确定量检测真菌毒素的主要手段,其优势在于该方法具有灵敏度高、定量准确和抗干扰能力强等特点。传统的样品前处理净化策略(例如固相萃取柱净化和免疫亲和柱净化)往往仅适用于一种或特定类型的毒素化合物[12]。已有文献[13-16]报道采用直接稀释-液相色谱-串联质谱法同时检测食品中多种真菌毒素化合物,这些方法往往需要超过20 min的色谱分析时间,或仅覆盖十种甚至更少的毒素种类。本试验旨在建立一种适用于多种食品基质的多真菌毒素快速检测方法,以更短的色谱分析时间,定量分析涵盖我国现行真菌毒素限量标准[5](GB 2761—2017)以及欧盟法规[2]中涉及的真菌毒素及其相关衍生物,从而更高效、全面地提供我国食品中真菌毒素污染情况信息。
本试验全面优化前处理以及色谱、质谱各环节技术参数,建立适用于常规实验室应用的同位素内标定量检测粮谷类食品中16种真菌毒素的方法,可为我国食品中多真菌毒素风险监测与暴露评估提供技术支持。
1材料与方法
1.1材料
1.1.1样品来源
30份谷物及其制品样品包括15份玉米粉、5份小麦粉、5份方便面和5份面包,均购自杭州市某农贸市场和超市。
1.1.2主要仪器与试剂
超高效液相色谱-质谱联用仪(美国Waters)、电子分析天平、高速冷冻离心机、氮吹仪、多孔涡旋振荡器、试验筛(0.5~1 mm)、高速粉碎机。AFTB1(CAS:1162-65-8)、AFTB2(CAS:7220-81-7)、AFTG1(CAS:1165-39-5)、AFTG2(CAS:7241-98-7)、雪腐镰刀菌烯醇(NIV,CAS:023282-20-4)、DON(CAS:51481-10-8)及其乙酰化衍生物(3-ADON,CAS:50722-38-8;15-ADON,CAS:88337-96-6)、ZEN(CAS:017924-92-408)、OTA(CAS:000303-47-9)、FB1(CAS:116355-83-0)、FB2(CAS:116355-84-1)、FB3(CAS:136379-59-4)、T-2毒素(CAS:021259-20-1)、HT-2毒素(CAS:026934-87-2)和杂色曲霉素(ST,CAS:10048-13-2)16种真菌毒素标准品以及相应的15种13C同位素内标标准溶液(纯度≥98%,无15-ADON的同位素内标标准溶液)均购自奥地利RomeLab,乙腈、甲醇、甲酸均为色谱纯,试验用水为超纯水,玉米粉参考物质[T2298QC和T4230QC,英国弗帕斯检测技术研究所(FAPAS)]。
1.2方法
1.2.1标准溶液的制备
标准储备液:用乙腈分别溶解或稀释DON、NIV、ZEN、3-ADON、15-ADON、HT-2、T-2等标准品至100 μg/ml,用乙腈分别溶解或稀释AFTB1、AFTB2、AFTG1、AFTG2、OTA等标准品至1 μg/ml,用乙腈稀释ST标准溶液至10 μg/ml,得到相应标准储备液;用50%乙腈-水溶液分别稀释FB1、FB2、FB3等标准品至50 μg/ml,得到相应标准储备液。混合标准储备液:取各单一标准储备液2 ml于10 ml容量瓶,用乙腈定容至刻度,即得混合标准储备液。
混合同位素内标工作液:分别移取一定体积的同位素标准溶液,用乙腈稀释定容,得到混合同位素内部工作液,其中13C-AFTB1、13C-AFTB2、13C-AFTG1、13C-AFTG2、13C-OTA含量为0.01 μg/ml,13C-NIV、13C-DON、 13C-3ADON、13C-ZEN含量为1.25 μg/ml,13C-ST含量为0.02 μg/ml,13C-T-2含量为0.02 μg/ml,13C-HT-2、 13C-FB1、13C-FB2、13C-FB3含量为0.5 μg/ml。
标准曲线溶液:准确移取混合标准储备液适量,采用20%乙腈-水溶液逐级稀释,配制成分别含AFTB1 0.05、0.1、0.2、0.5、1.0、2.0 ng/ml的标准曲线系列溶液。移取20 μl同位素内标混合溶液于内插管中,加入180 μl对应的混合标准曲线浓度点溶液,混匀即得标准曲线溶液系列。
1.2.2样品制备
谷物及其制品(至少1 kg)用高速粉碎机将其粉碎,过筛,使其粒径小于1 mm孔径试验筛,混合均匀后缩分至100 g,储存于样品瓶中,密封保存,供检测用。
1.2.3样品提取与净化
称取5 g样品于50 ml离心管中,加入20 ml乙腈-水-甲酸(79∶20∶1,V/V)溶液,并用涡旋混合器混匀1 min,置于旋转摇床上振荡提取30 min,取1.0 ml提取液至1.5 ml离心管中,以10 000 r/min离心5 min。准确转移0.5 ml上清液于另一1.5 ml离心管中,加入1.0 ml水,旋涡混匀后,在4 ℃下以10 000 r/min离心5 min,吸取上清液过0.22 μm滤膜。吸取180 μl处理好的样品滤液于300 μl内插管中,加入20 μl稳定同位素混合溶液,涡旋混匀,待进样。
1.2.4仪器条件
色谱:Waters Cortecs UPLC C18柱(100 mm×3.0 mm,1.6 μm),流动相以0.1%甲酸溶液为水相(A),乙腈作为有机相(B),线性梯度洗脱:0~0.8 min 10%B,0.8~2.5 min 28%B,2.5~8 min 45%B,8~8.5 min 68%B,8.5~11.5 min 68%B,11.5~12 min 100%B,12~13 min 100%B,13~13.5 min 10%B,进样量为5 μl。质谱:电喷雾离子源(ESI),毛细管电压2.5 kV(ESI+)/3.5 kV(ESI-),离子源温度为150 ℃,脱溶剂气温度为500 ℃,脱溶剂气流速为800 L/h,多反应监测模式(MRM)。
2结果与分析
2.1超高效液相色谱-串联质谱条件的选择及优化
首先考察16种真菌毒素的离子化模式。将各毒素化合物的标准储备液用乙腈稀释定容至1 μg/ml,通过色谱柱-质谱进样法,分别评估正、负离子模式下该16种毒素化合物的电离情况。结果显示,在正离子模式下,所有的化合物(除NIV外)均有较好的响应,而在负离子模式下,仅NIV、DON、3-ADON、15-ADON和ZEN这5种化合物有响应,因此,除NIV外的其他15种化合物采用正离子模式优化最佳母离子和子离子参数,NIV采用负离子模式优化质谱条件参数,16种真菌毒素化合物的质谱参数如表1所示。
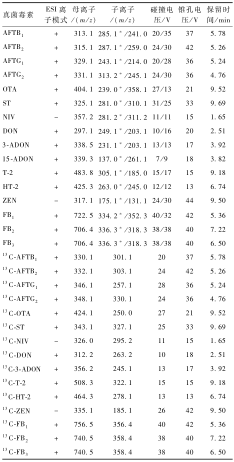
|
表116种真菌毒素的质谱参数 Table 1Mass spectrum parameters of 16 compounds of mycotoxins 注:*表示定量离子通道 |
流动相的差异会直接影响到目标化合物的离子化效率。根据文献[9,13,15-16]报道以及各化合物的分子结构特征,乙酸铵或甲酸铵的加入可以抑制加钠峰[M+Na]+的形成,酸的加入有利于改善如FBs这一类酸性毒素的峰型;因此,本研究考察了甲酸溶液(0.1%)、乙酸铵溶液(5 mmol/L)、甲酸(0.1%)-乙酸铵溶液(5 mmol/L),以及乙腈、甲醇不同有机溶剂对各化合物的离子化效率的影响。如图1所示,采用甲醇作为有机相时,大部分单端孢霉烯族类毒素(如DON、3-ADON、15-ADON以及HT-2)响应灵敏度较高,而其他的毒素,特别是AFTs、OTA这两类限量值较低的毒素,在乙腈作为流动相时,响应灵敏度较甲醇条件下的高。甲醇的洗脱能力弱于乙腈,当用甲醇作为强洗脱溶剂时,整个色谱分离时间会相对较长。从图1中还可以看到,5 mmol/L的乙酸铵仅可以增强T-2和ST毒素的响应,但0.1%甲酸作为水相溶剂时,其他大部分毒素都有较好的响应。为了保证目标分析物的响应最佳和缩短分析时间,本研究最终选择0.1%甲酸水-乙腈溶液为流动相。
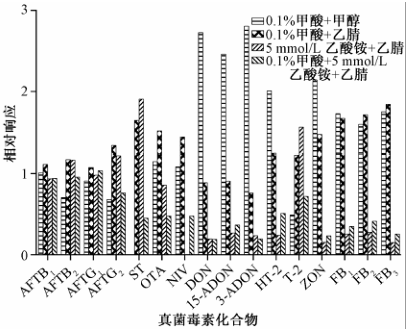
|
图1流动相对16种真菌毒素离子化效应的影响 Figure 1Effect of mobile phase on ionization of 16 mycotoxin compounds |
2.2前处理条件的优化
多真菌毒素的检测方法,其关键步骤为毒素的提取和净化。为了尽可能的减少前处理过程中毒素的损失并提高毒素提取的种类,本试验全面考察前处理过程中各环节的影响因素,以玉米粉为首要考察对象,优化建立了一种快捷简单的一步提取净化法。甲醇和乙腈水溶液体系是真菌毒素提取过程中最常用的有机溶剂,但实际试验中显示乙腈对蛋白沉淀效果以及提取液离心后的澄清度均优于甲醇,故本试验对不同比例的乙腈-水溶液进行了考察。图2结果表明,对大多数毒素化合物,提取溶液有机相比例在70%~90%之间,均有较好的回收率。亲水性较强的如呕吐类毒素,提取溶液水相比例高时,提取回收率最佳;而亲脂性较强的如ZEN化合物,提取溶液有机相比例高时,提取回收率最佳。综合考虑16种真菌毒素的最佳平均提取回收率,故本试验采用80%乙腈-水溶液作为提取溶液。
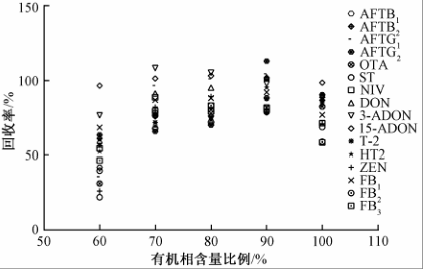
|
图2提取液有机相比例对16种真菌毒素提取回收率的影响 Figure 2Effect of organic phase in extraction solvent for 16 mycotoxins recoveries |
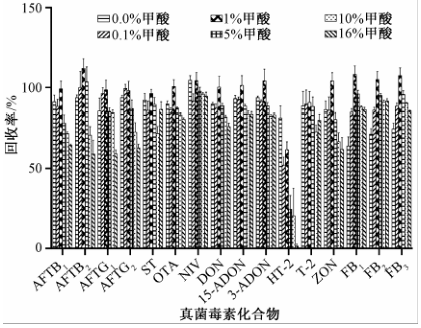
|
图3提取液中甲酸含量对16种真菌毒素提取效率的影响 Figure 3Effect of formic acid in extraction solvent for 16 mycotoxins recoveries |
2.3方法学验证
本试验将优化完善后建立的直接稀释-液相色谱-串联质谱法应用于玉米粉、小麦粉、方便面和面包等4种常见食品基质,并分别对该4种食品基质开展本方法的方法学验证。
2.3.1基质效应的评价
基质效应(matrix effect,ME)是指样品基质中的内源性物质会在离子源(尤其是ESI源)中与待测物质竞争发生离子化反应,导致响应下降或升高的现象,这是液相色谱-质谱法在测定实际样品时无可避免的现象。鉴于真菌毒素污染的普遍性,很难获得完全空白的基质,同时同位素内标与目标化合物具有极其相似的色谱质谱行为,故本试验采用提取液加入同位素内标法,评估玉米粉、小麦粉、方便面和面包等4种食品基质的ME。选取相应的空白基质样品加入15种混合同位素内标标准溶液,混匀后分析,比较各同位素内标化合物在基质溶液与在溶剂中的响应峰面积比值。比值越接近于1,说明ME越小,反之,ME越严重。表2结果显示,16种真菌毒素的ME在27.3%~281.4%,说明在这4种食品基质中存在一定的基质增强或抑制效应。基质匹配标准曲线是一种较为常见的校准ME的方法。但真菌毒素污染具有普遍性,完全不含16种真菌毒素化合物的空白基质不易获取。同时,加工工艺的不同会导致样品基质的不一致性,代表性基质同样不易获取,因此,为了更好地消除ME带来的定量影响,本试验采用同位素稀释内标法定量,以确保分析结果的准确可靠。
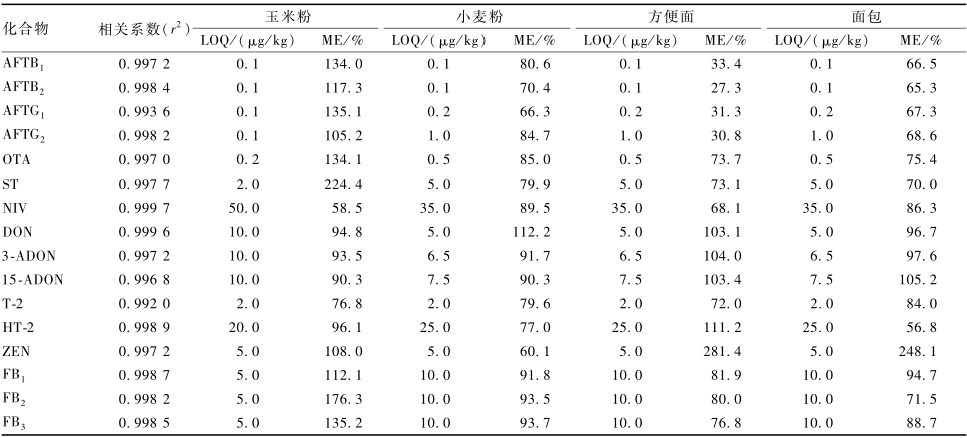
|
表216种真菌毒素在不同食品基质中的线性、ME与方法定量限(LOQ) Table 2Linearity, matrix effect and LOQ of 16 mycotoxins in different food matrices |
2.3.2线性范围及定量限
配制16种真菌毒素混合标准储备液,用20%乙腈-水溶液逐级稀释,配成系列混合标准溶液,以同位素稀释法定量,结果如表2所示。在各目标化合物的线性范围内,16种真菌毒素线性关系良好,r2>0.99。分别选取空白的玉米粉、小麦粉、方便面和面包作为考察对象,加入适量16种真菌毒素混合标准溶液,进行提取净化,分离检测。以3倍信噪比(S/N=3)和10倍信噪比(S/N=10)时相应的加标量为方法的检出限(LOD)与LOQ,结果见表2。16种真菌毒素LOQ均低于我国和欧盟真菌毒素限量法规[2,5],本方法可以满足日常监测需要。
2.3.3回收率与精密度
分别选取空白的玉米粉、小麦粉、方便面和面包作为考察对象,加入低、中、高3个浓度水平的16种真菌毒素混合标准溶液[17],进行提取净化,分离检测。每个加标水平3次平行试验,加标浓度、回收率以及精密度见表3。加标回收率在72.67%~126.92%之间,相对标准偏差(RSD)为0.15%~16.83%,符合欧盟法规对于多真菌毒素检测方法回收率和RSD的要求[18]。
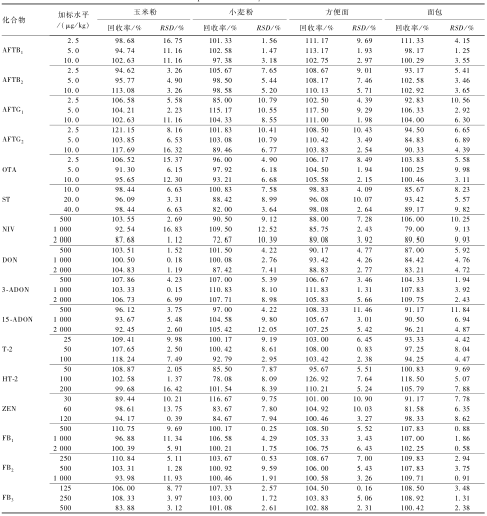
|
表316种真菌毒素在不同食品基质中的回收率与精密度(n=3) Table 3Recoveries and precious of 16 mycotoxins in different food matrices |
2.3.4准确度
为进一步验证方法的准确性和适用性,本试验测定了FAPAS能力验证玉米样品(T2298QC和T04230QC),结果如表4所示。结果显示,目标化合物测定值均在标示范围内,且接近标示中位值,表明本方法准确可靠。
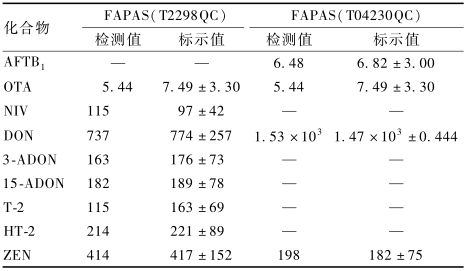
|
表4FAPAS质控样品检测结果(±s,μg/kg) Table 4Validation results of mycotoxins in FAPAS quality control samples 注:—表示质控样品证书中未列出该项标示值 |
2.4方法应用与实际样品检测
采用优化后方法对30份玉米粉、小麦粉、方便面和面包样品进行检测。结果显示,共检出NIV、DON、3-ADON、15-ADON、AFTB1、OTA、ZEN、ST、FB1、FB2以及FB3等11种真菌毒素,其他5种均未检出。玉米样品中共有10种真菌毒素检出,其中AFTB1、OTA和ST检出含量较低,DON和FBs含量分布范围较广。而小麦粉、面包和方便面则以DON和NIV两种真菌毒素污染为主,其他化合物检出较少。这一方面与真菌易感植株体种类有着密切关系,另一方面本试验所选面包和方便面样品均为小麦粉制品,其所含真菌毒素种类与小麦粉中所含毒素种类有着极高的相似度。从结果还可以看出,不论玉米粉还是小麦粉样品,DON含量分布均较为广泛。这可能与农作物种植地温湿度以及收割后运输、储藏环境有关系。根据GB 2761—2017《食品安全国家标准 真菌毒素限量标准》[5]中谷物及其制品中DON限量为1 000 μg/kg的规定,15份玉米粉样品DON含量检出范围为27~596 μg/kg,无超标样品。而15份小麦粉及其制品中DON含量检出范围为115~1 673 μg/kg,其中有2份小麦粉样品和2份方便面样品超过限量值。
3小结
本试验采用一步提取净化法,结合同位素稀释法,建立了快速准确地定量分析谷物及其制品中16种真菌毒素的液相色谱-串联质谱方法。本方法对前处理条件、色谱及质谱条件作了全面且细致地优化,并采用加标回收及基体标准物质验证了本方法的灵敏度和准确度。本方法缩短了色谱分析时间,提高检测方法的环境友好性;精简了前处理步骤,避免了前处理过程中目标化合物的损失与误差。基于三重四极杆的检测方法,本方法既符合我国和欧盟真菌毒素限量法规要求,同时可适用与谷物及其制品中多真菌毒素的测定,可满足常规实验室快速、准确定量分析谷类食品中真菌毒素污染情况的需求。
参考文献
[1]EGMOND H, JONKER M A. Worldwide regulations for mycotoxins in food and feed in 2003 [M]. Food Agric Organiz United Nations, 2004.
[2]Commission Regulation (EU) NO 1881/2006 setting maximum levels for certain contaminants in foodstuffs [S]. Official Journal of the European Union, 2006.
[3]Codex Alimentarius Commission (CAC). Commission CODEX STAN 193-1995 codex general standard for contaminants and toxins in food and feed [M]. 1995.
[4]RYCHLIK M, LEPPER H, WEIDNER C, et al. Risk evaluation of the alternaria mycotoxin tenuazonic acid in foods for adults and infants and subsequent risk management [J]. Food Control,2016, 68(3):181-185.
[5]中华人民共和国国家卫生和计划生育委员会,国家食品药品监督管理总局.食品安全国家标准 食品中真菌毒素限量:GB 2761—2017 [S]. 北京:中国标准出版社,2017.
[6]GORYACHEVA I Y, SAEGER S D, EREMIN S A, et al. Immunochemical methods for rapid mycotoxin detection: evolution from single to multiple analyte screening: a review [J]. Food Additives and Contaminants,2007, 24(10): 1169-1183.
[7]XU J J, ZHOU J, HUANG B F, et al. Simultaneous and rapid determination of deoxynivalenol and its acetylate derivatives in wheat flour and rice by ultra high performance liquid chromatography with photo diode array detection [J]. Journal of Separation Science, 2016, 39(11): 2028-2035.
[8]ZHOU J, XU J J, HUANG B F, et al. High-performance liquid chromatographic determination of multi-mycotoxin in cereals and bean foodstuffs using interference-removal solid-phase extraction combined with optimized dispersive liquid-liquid microextraction [J]. Journal of Separation Science, 2017, 40(10): 2141-2150.
[9]REN Y, ZHANG Y, SHAO S, et al. Simultaneous determination of multi-component mycotoxin contaminants in foods and feeds by ultra-performance liquid chromatography tandem mass spectrometry [J]. Journal of Chromatography A, 2007, 1143(1/2): 48-64.
[10]JIN P G, HAN Z, CAI Z X, et al. Simultaneous determination of 10 mycotoxins in grain by ultra-high-performance liquid chromatography-tandem mass spectrometry using 13C15-deoxynivalenol as internal standard [J]. Food Additives and Contaminants: Part A, 2010, 27(12): 1701-1713.
[11]BREIDBACH A. A greener, quick and comprehensive extraction approach for LC-MS of multiple mycotoxins [J]. Toxins, 2017, 9(3): 91-114.
[12]PEREIRA V L, FERNANDES J O, CUNHA S C. Comparative assessment of three cleanup procedures after QuEChERS extraction for determination of trichothecenes (type A and type B) in processed cereal-based baby foods by GC-MS [J]. Food Chemistry, 2015, 182(1): 143-149.
[13]RASMUSSEN R R, STORM I M L D, RASMUSSEN P H, et al. Multi-mycotoxin analysis of maize silage by LC-MS/MS [J]. Analytical and Bioanalytical Chemistry, 2010, 397(2): 765-776.
[14]OUESLATI S, ROMERO-GONZLEZ R, LASRAM S, et al. Multi-mycotoxin determination in cereals and derived products marketed in Tunisia using ultra-high performance liquid chromatography coupled to triple quadrupole mass spectrometry [J]. Food and Chemical Toxicology,2012, 50(7): 2376-2381.
[15]ZHU R, ZHAO Z Y, WANG J H, et al. A simple sample pretreatment method for multi-mycotoxin determination in eggs by liquid chromatography tandem mass spectrometry [J]. Journal of Chromatography A, 2015, 1417(9): 1-7.
[16]FLORES-FLORES M E, GONZLEZ-PEAS E. An LC-MS/MS method for multi-mycotoxin quantification in cow milk [J]. Food Chemistry, 2017, 218(9): 378-385.
[17]U.S. FDA. Guidelines for the validation of analytical methods for the detection of microbial pathogens in foods and feeds [M]. 2nd Edition, 2015.
[18]Commission Regulation (EU) No 519/2014 of 16 May 2014 amending regulation (EC) No 401/2006 as regards methods of sampling of large lots, spices and food supplements, performance criteria for T-2, HT-2 toxin and citrinin and screening methods of analysis [S]. Official Journal of the European Union, 2014.
[2]Commission Regulation (EU) NO 1881/2006 setting maximum levels for certain contaminants in foodstuffs [S]. Official Journal of the European Union, 2006.
[3]Codex Alimentarius Commission (CAC). Commission CODEX STAN 193-1995 codex general standard for contaminants and toxins in food and feed [M]. 1995.
[4]RYCHLIK M, LEPPER H, WEIDNER C, et al. Risk evaluation of the alternaria mycotoxin tenuazonic acid in foods for adults and infants and subsequent risk management [J]. Food Control,2016, 68(3):181-185.
[5]中华人民共和国国家卫生和计划生育委员会,国家食品药品监督管理总局.食品安全国家标准 食品中真菌毒素限量:GB 2761—2017 [S]. 北京:中国标准出版社,2017.
[6]GORYACHEVA I Y, SAEGER S D, EREMIN S A, et al. Immunochemical methods for rapid mycotoxin detection: evolution from single to multiple analyte screening: a review [J]. Food Additives and Contaminants,2007, 24(10): 1169-1183.
[7]XU J J, ZHOU J, HUANG B F, et al. Simultaneous and rapid determination of deoxynivalenol and its acetylate derivatives in wheat flour and rice by ultra high performance liquid chromatography with photo diode array detection [J]. Journal of Separation Science, 2016, 39(11): 2028-2035.
[8]ZHOU J, XU J J, HUANG B F, et al. High-performance liquid chromatographic determination of multi-mycotoxin in cereals and bean foodstuffs using interference-removal solid-phase extraction combined with optimized dispersive liquid-liquid microextraction [J]. Journal of Separation Science, 2017, 40(10): 2141-2150.
[9]REN Y, ZHANG Y, SHAO S, et al. Simultaneous determination of multi-component mycotoxin contaminants in foods and feeds by ultra-performance liquid chromatography tandem mass spectrometry [J]. Journal of Chromatography A, 2007, 1143(1/2): 48-64.
[10]JIN P G, HAN Z, CAI Z X, et al. Simultaneous determination of 10 mycotoxins in grain by ultra-high-performance liquid chromatography-tandem mass spectrometry using 13C15-deoxynivalenol as internal standard [J]. Food Additives and Contaminants: Part A, 2010, 27(12): 1701-1713.
[11]BREIDBACH A. A greener, quick and comprehensive extraction approach for LC-MS of multiple mycotoxins [J]. Toxins, 2017, 9(3): 91-114.
[12]PEREIRA V L, FERNANDES J O, CUNHA S C. Comparative assessment of three cleanup procedures after QuEChERS extraction for determination of trichothecenes (type A and type B) in processed cereal-based baby foods by GC-MS [J]. Food Chemistry, 2015, 182(1): 143-149.
[13]RASMUSSEN R R, STORM I M L D, RASMUSSEN P H, et al. Multi-mycotoxin analysis of maize silage by LC-MS/MS [J]. Analytical and Bioanalytical Chemistry, 2010, 397(2): 765-776.
[14]OUESLATI S, ROMERO-GONZLEZ R, LASRAM S, et al. Multi-mycotoxin determination in cereals and derived products marketed in Tunisia using ultra-high performance liquid chromatography coupled to triple quadrupole mass spectrometry [J]. Food and Chemical Toxicology,2012, 50(7): 2376-2381.
[15]ZHU R, ZHAO Z Y, WANG J H, et al. A simple sample pretreatment method for multi-mycotoxin determination in eggs by liquid chromatography tandem mass spectrometry [J]. Journal of Chromatography A, 2015, 1417(9): 1-7.
[16]FLORES-FLORES M E, GONZLEZ-PEAS E. An LC-MS/MS method for multi-mycotoxin quantification in cow milk [J]. Food Chemistry, 2017, 218(9): 378-385.
[17]U.S. FDA. Guidelines for the validation of analytical methods for the detection of microbial pathogens in foods and feeds [M]. 2nd Edition, 2015.
[18]Commission Regulation (EU) No 519/2014 of 16 May 2014 amending regulation (EC) No 401/2006 as regards methods of sampling of large lots, spices and food supplements, performance criteria for T-2, HT-2 toxin and citrinin and screening methods of analysis [S]. Official Journal of the European Union, 2014.