 梁素丹,陈剑刚,张艳.全自动固相萃取-超高效液相色谱-串联质谱法同时测定动物肌肉中喹诺酮类和四环素类兽药残留[J].中国食品卫生杂志,2018,30(2):151-157.
梁素丹,陈剑刚,张艳.全自动固相萃取-超高效液相色谱-串联质谱法同时测定动物肌肉中喹诺酮类和四环素类兽药残留[J].中国食品卫生杂志,2018,30(2):151-157. DOi:10.13590/j.cjfh.2018.02.006
全自动固相萃取-超高效液相色谱-串联质谱法同时测定动物肌肉中喹诺酮类和四环素类兽药残留
(珠海市疾病预防控制中心,广东 珠海519000)
收稿日期:2018-03-26
作者简介:梁素丹女副主任技师研究方向为食品违禁药物及环境污染物分析E-mail:lsudan@126.com
通信作者:陈剑刚男主任技师研究方向为食品违禁药物及环境污染物分析E-mail:davidchenjg@sina.com
基金项目:
摘要:目的建立同时测定肌肉中氧氟沙星、诺氟沙星、培氟沙星、环丙沙星、达氟沙星、洛美沙星、恩诺沙星、二氟沙星、沙拉沙星、噁喹酸和氟甲喹等11种喹诺酮类和四环素、金霉素、土霉素和强力霉素等4种四环素类药物残留的全自动固相萃取-超高效液相色谱-串联四级杆质谱分析方法。方法肌肉(包括鸡肉、猪肉、虾肉)以含0.1 mol/L EDTA-Mcllcaine缓冲溶液(pH=4.0)提取,HLB固相萃取柱净化,100%甲醇洗脱。采用UPLC C18色谱柱(100 mm×2.1 mm,1.6 μm)分离,以0.1%甲酸水和甲醇-乙腈(40∶60,V/V)溶液为流动相进行梯度洗脱,采用电喷雾正离子电离,多反应监测(MRM)模式,以保留时间和目标物的二级质谱特征碎片离子予以双定性确证,以基质外标法定量。结果15个组分与杂质能得到良好分离,在1.25~50.0 μg/kg范围内线性关系良好(r≥0.998 5),11种喹诺酮类药物最低检出限和定量限分别为0.013~0.069和0.043~0.23 μg/kg,4种四环素类药物最低检出限和定量限分别为0.033~0.093和0.11~0.31 μg/kg。高、中、低三种添加水平的加标回收试验:鸡肉的加标回收率为70.8%~105.4%,相对标准偏差为0.5%~7.4%;猪肉的加标回收率为75.6%~115.2%,相对标准偏差为0.8%~8.9%;虾肉的加标回收率为73.7%~117.5%,相对标准偏差为0.5%~14.8%。应用该方法对广东省60份实际样品进行检测,结果在4份样品中分别检出氧氟沙星、环丙沙星、恩诺沙星和强力霉素,其中虾肉样品的恩诺沙星和强力霉素含量均高达400 μg/kg,鸡肉样品中强力霉素含量高达220 μg/kg,均超出限量值(100 μg/kg)。结论本方法准确、快捷、简便,适用于鸡肉、猪肉和虾肉等动物肌肉中喹诺酮类和四环素类的同步确证及定量分析,为食品安全风险监测提供高效可靠的方法。
关键词:
超高效液相色谱-串联四级杆质谱; 喹诺酮; 四环素; 全自动固相萃取; 肉; 兽药残留; 食品污染物; 测定
文章编号:1004-8456(2018)02-0151-07
中图分类号:R155
文献标志码:A
Simultaneous determination of veterinary drug residues of quinolones and tetracyclines in animal tissue by ultra performance liquid chromatography-tandem mass spectrometry with automatic solid phase extraction
(Zhuhai Center for Disease Control and Prevention,Guangdong Zhuhai 519000,China)
Abstract:ObjectiveTo establish a method for the simultaneous determination of ofloxacin, norfloxacin, ciprofloxacin, pefloxacin, danofloxacin, lomefloxacin, enrofloxacin, ciprofloxacin, pefloxacin, oxolinic acid and flumequine,tetracycline, chlortetracycline, oxytetracycline and doxycycline in animal tissue by ultra performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) with automatic solid phase extraction. MethodsThe samples were extracted with 0.1 mol/L EDTA-Mcllcaine buffer solution (pH=4.0), and the resulting extracts were cleaned-up on HLB solid phase extraction column, and then eluted with 100% methanol. The target components were separated on a UPLC C18 column (100 mm×2.1 mm, 1.6 μm), with 0.1% formic acid water and methanol-acetonitrile (40∶ 60, V/V) solution as mobile phase in gradient elution. Multiple reaction monitoring (MRM) in positive was used, qualitative confirmation was performed from retention time and secondary mass characteristic ions, and the matrix-matched external standard calibration curves were used for quantitative analysis. ResultsThe target components and impurity could be well separated, it was showed good linearity in the range of 1.25-50.0 μg/kg (r=0.998 5 above). The detection limits and quantitation limits of 11 kinds of quinolones were 0.013-0.069 and 0.043-0.23 μg/kg, the limits of 4 kinds of tetracyclines were 0.033-0.093 and 0.11-0.31 μg/kg. The recovery tests at high, medium, and low spiked levels: the recoveries were 70.8%-105.4%, and relative standard deviation (RSDs) were 0.5%-7.4% in chicken; the recoveries were 75.6%-115.2%, and RSDs were 0.8%-8.9% in pork; the recoveries were 73.7%-117.5%, and RSDs were 0.5%-14.8% in shrimp. The method was applied to the analysis of 60 samples from Guangdong, and the result showed that ofloxacin, ciprofloxacin, enrofloxacin and doxycycline were detected in 4 samples, the maximum content of enrofloxacin and doxycycline in shrimp was 400 μg/kg, the content of doxycycline in chicken was 220 μg/kg, which exceeded the limit value of 100 μg/kg. ConclusionThe method was accurate, efficient and simple, synchronous confirmation and quantitative analysis of the residues for chicken, pork and shrimp meat and other musculature of quinolones and tetracyclines, and was suitable for analysis and confirmation in food safety risk monitoring.
Key words:
Ultra performance liquid chromatography-tandem mass spectrometry; quinolones; tetracyclines; automated solid phase extraction; meat; veterinary drug residues; food contaminants; determination
喹诺酮类(quinolones,QNs)和四环素类(tetracyclines,TCs)药物是广谱抗生素,大多数是人畜共用药物,如四环素、土霉素、恩诺沙星、环丙沙星等。由于在动物饲养过程中的不科学使用甚至滥用,导致在动物源性食品中的残留,并随着食物链进入人体,诱发致病菌产生耐药性,QNs药物还有潜在的致癌性和遗传毒性,威胁着人类的健康。同时两类药物作为新型污染物(PPCPs),已引起了社会的广泛关注[1-3],我国以及世界卫生组织、欧盟、美国、日本等国家和组织都将QNs和TCs列入限制使用的兽药名单中,并制定出相关的最高残留量(MRL)。
目前对QNs和TCs的检测方法主要有酶联免疫(ELISA)法[4-5]、微生物法[6]、高效液相色谱(HPLC)法[7-9]和超高效液相色谱-串联质谱(UPLC-MS/MS)法[10-12]。ELISA法和微生物法特异性不强,假阳性率高;HPLC法特异性强、定量准确,但是其抗干扰能力差,故定性能力也差;UPLC-MS/MS法具有选择性强、灵敏度高、检出限低、集高效分离和结构鉴定为一体,已成为复杂混合物中痕量组分定性和定量分析的有力工具。能同时提取并净化肌肉中这两类药物多种残留的研究方法不多[12-14],几乎都是针对单一的鸡肉或猪肉,而且研究的药物种类在当今的食品安全监控中是远远不够的。本试验选择鸡肉、猪肉和虾肉3种肌肉中11种QNs及4种TCs药物为研究对象,统一对两类物质的前处理过程,并用UPLC-MS/MS同时分析,有效的缩短了检测时间,为食品安全风险监测中QNs和TCs药物的分析和监测提供可靠的方法。
11种QNs和4种TCs药物标准品均购自德国Dr.Ehrenstonfer,纯度均>95%(见表1)。乙腈、甲醇均为高效液相级,甲酸(色谱级),柠檬酸、十二水合磷酸氢二钠、乙二胺四乙酸二钠均为分析纯,超纯水(18.2 MΩ·cm)。乙二胺四乙酸(EDTA)-Mcllcaine缓冲液:分别称取12.9 g柠檬酸、10.9 g磷酸氢二钠、37.2 g乙二胺四乙酸二钠,加水溶解并定容至1 000 ml,用氢氧化钠调节pH值至(4.0±0.05)。
质谱:电喷雾离子源,正离子模式(ESI+)分段采集(0.00~5.20,5.20~12.00 min),多反应监测(MRM)。毛细管电压3.5 kV,锥孔电压30 V,离子源温度150 ℃,脱溶剂温度500 ℃。其他参数见表3。
QNs和TCs均为酸碱两性化合物,其离解状态和在流动相中的溶解性随流动相的pH值而变化,因此本试验采用0.1%甲酸水控制流动相的pH值,有效改善峰型,加强离子化效应,提高灵敏度。并考察乙腈-0.1%甲酸水溶液及0.1%甲酸水溶液和甲醇-乙腈(40∶60,V/V)溶液两种流动相对分析物离子化效率和色谱峰形的影响。虽然质谱分析不要求在色谱上完全分离,但QNs药物中存在母离子和子离子均相同的化合物,如氟甲喹和噁喹酸均具有262.2>244.0离子对,必须要求在色谱上分离,否则会互相干扰。试验发现,流动相为0.1%甲酸水溶液和甲醇-乙腈(40∶60,V/V)溶液时,对于灵敏度较低的金霉素和强力霉素峰型更加对称和尖锐,调整梯度洗脱(见表2),氟甲喹和噁喹酸能完全分离。
HLB小柱是兼具亲脂和亲水基团的反相柱,对极性化合物具有优异的保留能力。净化处理选择全自动固相萃取装置进行,前提是提取液必须经低温离心(<5 ℃),使脂肪析出,并用少量脱脂棉球过滤去除脂肪和悬浮物后上样,避免固相萃取管路被堵塞而引起仪器故障。此装置可以同时处理44份样品,全程耗时约3.5 h,可实现全自动化固相萃取,减少时间和人力,其重复性比手动固相萃取更好,将是今后样品前处理的发展方向。
均在0.5 μg/kg以下,能满足国内食品安全风险监测的要求,可应用于动物肌肉中11种QNs和4种TCs药物的同步确证及定量分析。
目前对QNs和TCs的检测方法主要有酶联免疫(ELISA)法[4-5]、微生物法[6]、高效液相色谱(HPLC)法[7-9]和超高效液相色谱-串联质谱(UPLC-MS/MS)法[10-12]。ELISA法和微生物法特异性不强,假阳性率高;HPLC法特异性强、定量准确,但是其抗干扰能力差,故定性能力也差;UPLC-MS/MS法具有选择性强、灵敏度高、检出限低、集高效分离和结构鉴定为一体,已成为复杂混合物中痕量组分定性和定量分析的有力工具。能同时提取并净化肌肉中这两类药物多种残留的研究方法不多[12-14],几乎都是针对单一的鸡肉或猪肉,而且研究的药物种类在当今的食品安全监控中是远远不够的。本试验选择鸡肉、猪肉和虾肉3种肌肉中11种QNs及4种TCs药物为研究对象,统一对两类物质的前处理过程,并用UPLC-MS/MS同时分析,有效的缩短了检测时间,为食品安全风险监测中QNs和TCs药物的分析和监测提供可靠的方法。
1材料与方法
1.1材料
1.1.1试验材料
鸡肉、猪肉和淡水虾等样品从市场或超市购买,挑取可食用部分去筋、捣碎均匀。置于-18 ℃以下冰箱中储存备用。
1.1.2主要仪器与试剂
超高效液相色谱-串联四级杆质谱联用仪(I-class UPLC/Xevo-TQ-3)、HLB固相萃取柱(60 mg/3 ml)均购自美国Waters,低温高速离心机,漩涡混合器,Milli-Q超纯水系统,超声波清洗器,OA-SYS氮吹仪,全自动固相萃取仪(GX-274,美国吉尔森)。 11种QNs和4种TCs药物标准品均购自德国Dr.Ehrenstonfer,纯度均>95%(见表1)。乙腈、甲醇均为高效液相级,甲酸(色谱级),柠檬酸、十二水合磷酸氢二钠、乙二胺四乙酸二钠均为分析纯,超纯水(18.2 MΩ·cm)。乙二胺四乙酸(EDTA)-Mcllcaine缓冲液:分别称取12.9 g柠檬酸、10.9 g磷酸氢二钠、37.2 g乙二胺四乙酸二钠,加水溶解并定容至1 000 ml,用氢氧化钠调节pH值至(4.0±0.05)。
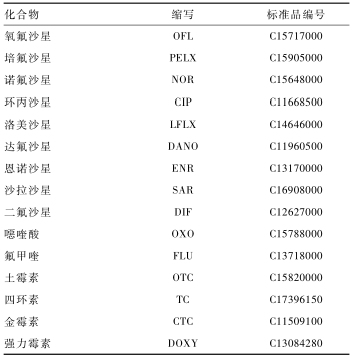
|
表1标准品信息 Table 1Standard information |
1.2方法
1.2.1样品前处理
准确称取2 g经绞碎均匀的动物肌肉样品于50 ml离心管中,加入10 ml 0.1 mol/L EDTA-Mcllcaine缓冲液,漩涡混合1 min,超声提取10 min,10 000 r/min离心5 min(温度低于5 ℃),分出上层溶液用脱脂棉球过滤,上清液备用。设置好全自动固相萃取装置参数,分别用3 ml甲醇活化、3 ml水平衡HLB柱。5 ml提取液以3 ml/min的速度过柱,弃去滤液,用2 ml 5%甲醇水溶液淋洗除杂质,真空抽干柱内液体后,分两次加入甲醇(3、2 ml)洗脱,收集洗脱液吹氮浓缩(40 ℃)至干,用1.0 ml 0.1%甲酸水溶解,漩涡混匀后过0.22 μm滤膜后待UPLC-MS/MS分析。
1.2.2标准溶液配制
分别精密称取QNs和TCs药物标准品0.010 0 g置于10 ml容量瓶中,用甲醇溶解并定容至刻度,即得1.0 mg/ml的单标贮备液,置于-20 ℃冰箱保存(注:噁喹酸贮备液配成浓度为0.5 mg/ml)。用甲醇分别逐级稀释配成1.0、0.1 μg/ml的混合标准应用液,用于基质标准曲线的配制。
1.2.3仪器条件
色谱:CORTECS UPLC C18色谱柱(100 mm×2.1 mm,1.6 μm),流动相:A为0.1%甲酸水溶液,B为甲醇-乙腈(40∶60,V/V)溶液,梯度洗脱(见表2),流速0.25 ml/min,柱温40 ℃,进样体积2 μl。
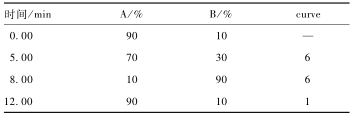
|
表2梯度洗脱程序 Table 2Gradient elution program for HPLC 注:curve为流动相梯度变化类型;—表示不设置 |
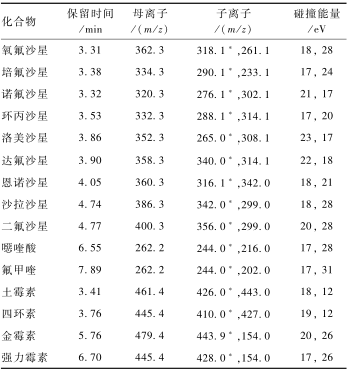
|
表311种QNs和4种TCs的质谱参数 Table 3Mass spectrometric parameters for 11 QNs and 4 TCs 注:*为定量离子 |
2结果与分析
2.1标准物质的溶解
本研究的15种药物最常用甲醇溶解标准物质并配制标准溶液,可是氧氟沙星、沙拉沙星和达氟沙星微溶于甲醇,尤其噁喹酸在甲醇中的溶解度<1.0%,它们为两性物质,易溶于酸或碱性溶液。而在众多学者的研究中并没有提及难溶的问题。在本实验标准贮备液配制时,除了噁喹酸需要降低浓度,加1~2滴氨水溶解后再用甲醇定容外,其他三组分选择了超声方式促溶解,进一步保证标准溶液配制的准确性。
2.2仪器条件选择
2.2.1色谱条件的选择
因QNs药物的叔氨基和羧基官能团能在水中发生离解,色谱柱固定相表面的残存硅醇基和金属离子可通过氢键或离子交换作用强烈吸附QNs化合物,出现色谱峰拖尾、保留时间不稳定或过长,甚至被保留在色谱柱上,导致峰型异常和分离下降。本试验选择CORTECS UPLC C18通用型高效反相色谱柱作为分析柱,利用亚2 μm实心核颗粒技术,在中、低pH值范围内可实现稳定的酸、碱和中性化合物保留性能,实现最高柱效。QNs和TCs均为酸碱两性化合物,其离解状态和在流动相中的溶解性随流动相的pH值而变化,因此本试验采用0.1%甲酸水控制流动相的pH值,有效改善峰型,加强离子化效应,提高灵敏度。并考察乙腈-0.1%甲酸水溶液及0.1%甲酸水溶液和甲醇-乙腈(40∶60,V/V)溶液两种流动相对分析物离子化效率和色谱峰形的影响。虽然质谱分析不要求在色谱上完全分离,但QNs药物中存在母离子和子离子均相同的化合物,如氟甲喹和噁喹酸均具有262.2>244.0离子对,必须要求在色谱上分离,否则会互相干扰。试验发现,流动相为0.1%甲酸水溶液和甲醇-乙腈(40∶60,V/V)溶液时,对于灵敏度较低的金霉素和强力霉素峰型更加对称和尖锐,调整梯度洗脱(见表2),氟甲喹和噁喹酸能完全分离。
2.2.2MRM质谱条件的选择
由于两类药物的结构和性质差异较大,测定时采用分组多通道采集,并分别对毛细管电压、锥孔电压、碰撞能量等质谱参数进行优化,采用正离子MRM模式,选择丰度较高的2个离子作为定量、定性特征离子,优化的质谱条件见表3,图1为空白鸡肉样品中添加11种QNs和4种TCs药物的总离子流图(TIC)及其特征离子色谱图(MRM)。
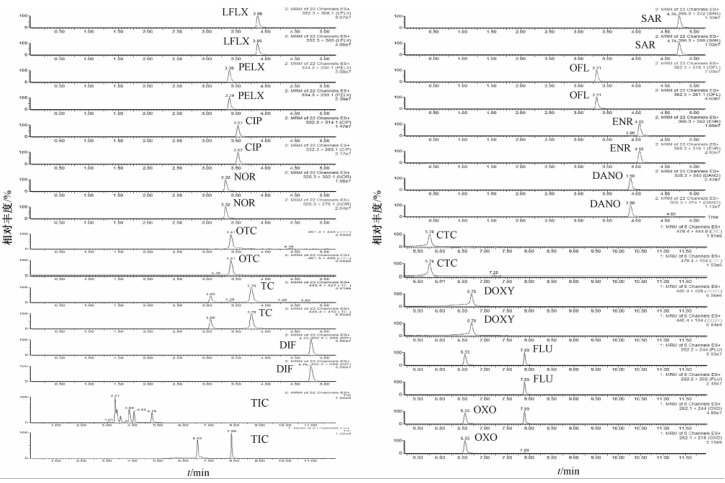
|
图1空白鸡肉加标样品的总离子流图(TIC)及其特征离子色谱图(MRM) Figure 1TIC and MRM chromatograms of a blank chicken with mixed standards |
2.3提取与净化条件的选择
QNs为极性化合物,易溶于极性和水溶性有机溶剂、稀酸和碱溶液,不溶于非极性溶剂。动物源性食品中QNs的提取剂大致可分为4种:1)水不溶性有机溶剂:如二氯甲烷;2)强极性有机溶剂:如乙腈、甲醇和乙酸乙酯;3)水溶性有机溶剂和酸、碱的混合液,如盐酸/磷酸/乙酸-乙腈等[10-11];4)缓冲溶液:如磷酸和柠檬酸缓冲溶液等[12-13]。而TCs物质容易与金属离子螯合形成沉淀,综合考虑,本试验用EDTA-Mcllcaine缓冲液作为提取剂,既可以掩蔽金属离子对TCs物质的影响,提供相应的pH值条件,又可以满足QNs物质的提取要求,更避免使用大量有机溶剂引起的污染。HLB小柱是兼具亲脂和亲水基团的反相柱,对极性化合物具有优异的保留能力。净化处理选择全自动固相萃取装置进行,前提是提取液必须经低温离心(<5 ℃),使脂肪析出,并用少量脱脂棉球过滤去除脂肪和悬浮物后上样,避免固相萃取管路被堵塞而引起仪器故障。此装置可以同时处理44份样品,全程耗时约3.5 h,可实现全自动化固相萃取,减少时间和人力,其重复性比手动固相萃取更好,将是今后样品前处理的发展方向。
2.4方法的线性范围、检出限
由于样品基质对待测组分测定影响较大,故采用基质工作曲线。称取鸡肉、猪肉和虾肉3种阴性样品2.0 g各6份,分别加入5、10、20、40、100、200 ng 15种组分的混合标准应用液,按1.2.1方法处理并取半量上净化柱,配制成1.25、2.5、5.0、10.0、25.0、50.0 μg/kg的基质工作曲线,目标化合物均具有良好的线性关系,相关系数均在0.998 5以上。按3倍信噪比和10倍信噪比计算方法最低检出限和定量限:11种QNs药物最低检出限和定量限分别为0.013~0.069和0.043~0.23 μg/kg,4种TCs药物最低检出限和定量限分别为0.033~0.093和0.11~0.31 μg/kg,结果见表4。
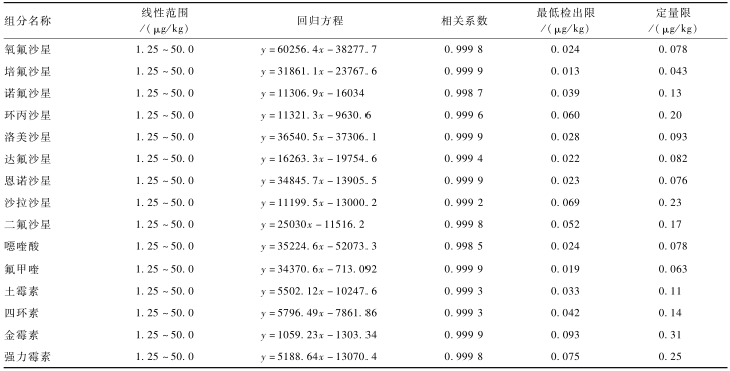
|
表411种QNs和4种TCs药物基质工作曲线的线性范围、回归方程、相关系数、最低检出限和定量限 Table 4Linear ranges, regression equations, correlation coefficients, LOD and LOQ of 11 QNs and 4 TCs |
2.5回收率和精密度
分别在阴性的鸡肉、猪肉、虾肉样品基质上添加10、20、40 μg/kg三种不同浓度水平的样品,每个浓度作5次重复测定。并以相应样品基质配制工作曲线进行校正分析,15个目标组分中鸡肉的加标回收率为70.8%~105.4%,相对标准偏差(RSD)为0.5%~7.4%;猪肉的加标回收率为75.6%~115.2%,RSD为0.8%~8.9%;虾肉的加标回收率为73.7%~117.5%,RSD为0.5%~14.8%,该方法具有较好的回收率和重现性,见表5。
2.6实际样品检测
应用该方法对2017年广东省4个地市区域随机抽查的60份样品(包括鸡肉、猪肉和虾肉)进行检测,结果在4份样品中分别检出氧氟沙星、环丙沙星、恩诺沙星和强力霉素,其中虾肉样品中恩诺沙星和强力霉素含量均高达400 μg/kg,鸡肉样品中强力霉素含量高达220 μg/kg,均超出限量值(100 μg/kg)。阳性样品色谱图见图2。
3小结
本试验建立了全自动固相萃取-UPLC-MS/MS法同时测定鸡肉、猪肉和虾肉等动物肌肉中11种QNs和4种TCs药物的残留分析方法,该方法简单、快速、准确,精密度和重现性好,15种药物的定量限
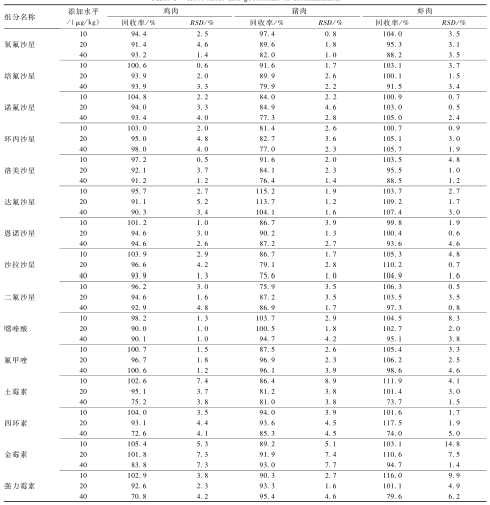
|
表511种QNs和4种TCs药物回收率和精密度试验结果(n=5) Table 5Recoveries and precisions of determination |

|
图2恩诺沙星(左)和强力霉素(右)阳性样品色谱图 Figure 2Chromatograms of positive samples of enrofloxacin (left) and doxycycline (right) |
参考文献
[1]姜利英,谢小品,崔光照,等.动物性食品中喹诺酮类药物残留检测方法的研究进展[J].郑州轻工业学院学报(自然科学版),2008,23(4):1-5.
[2]李慧芳,殷军港,刘永明.鱼肉中喹诺酮类药物残留量检测前处理方法的研究[J].中国渔业质量与标准,2012,2(1):62-66.
[3]刘峰,廖德润,李可,等.禽畜养殖基地磺胺类喹诺酮类和大环内酯类抗生素污染特征[J].农业环境科学学报,2013,32(4):847-853.
[4]沈洪刚,程玲玲,方智,等.酶联免疫快速测定猪肉中四环素族残留量[J].检验检疫科学,2000,10(2):42-43,54.
[5]刘智宏,叶妮,郭文林,等.四环素类药物多残留酶联免疫检测方法[J].中国农业科学,2009,42(1):318-323.
[6]黄晓蓉,郑晶,李寿菘,等.鳗鱼及其制品中喹诺酮类药物残留的微生物快速检测方法研究[J].淡水渔业,2005,35(4):3-5.
[7]赵思俊,李存,江海洋,等.高效液相色谱检测动物肌肉组织中7种喹诺酮类药物的残留[J].分析化学,2007,35(6):786-790.
[8]林玲,杨春亮,查玉兵,等.高效液相色谱法同时测定禽蛋中4种氟喹诺酮类药物残留量[J].分析仪器,2010(2):17-20.
[9]李娟.动物组织中11种喹诺酮类药物多残留的HPLC法研究[D].雅安:四川农业大学,2007.
[10]马建民,夏曦,李晓薇,等.阴离子交换固相萃取-超高效液相色谱-串联质谱法检测猪肌肉中13种喹诺酮类药物[J].中国食品卫生杂志,2013,25(3):249-253.
[11]刘柏林,谢继安,赵紫微,等.超高压液相色谱-电喷雾串联四极杆质谱内标法同时测定禽类食品中11种喹诺酮类药物[J].中国食品卫生杂志,2017,29(3):316-321.
[12]赵海香,孙艳红,丁明玉,等.多壁碳纳米管净化/超高效液相色谱串联质谱同时测定动物组织中四环素与喹诺酮多残留[J].分析测试学报,2011, 30(6):635-639.
[13]周鑫,李明,张鑫,等.高效液相色谱-串联质谱法测定鸡肉中的喹诺酮类和四环素类药物残留[J].畜牧与兽医,2015,47(11):19-22.
[14]倪永付,朱莉萍,王勇,等.微波辅助萃取/液相色谱-三重四极杆串联质谱法检测猪肉中氟喹诺酮类与四环素类药物残留[J].分析测试学报,2012,31(增刊):101-105.
[2]李慧芳,殷军港,刘永明.鱼肉中喹诺酮类药物残留量检测前处理方法的研究[J].中国渔业质量与标准,2012,2(1):62-66.
[3]刘峰,廖德润,李可,等.禽畜养殖基地磺胺类喹诺酮类和大环内酯类抗生素污染特征[J].农业环境科学学报,2013,32(4):847-853.
[4]沈洪刚,程玲玲,方智,等.酶联免疫快速测定猪肉中四环素族残留量[J].检验检疫科学,2000,10(2):42-43,54.
[5]刘智宏,叶妮,郭文林,等.四环素类药物多残留酶联免疫检测方法[J].中国农业科学,2009,42(1):318-323.
[6]黄晓蓉,郑晶,李寿菘,等.鳗鱼及其制品中喹诺酮类药物残留的微生物快速检测方法研究[J].淡水渔业,2005,35(4):3-5.
[7]赵思俊,李存,江海洋,等.高效液相色谱检测动物肌肉组织中7种喹诺酮类药物的残留[J].分析化学,2007,35(6):786-790.
[8]林玲,杨春亮,查玉兵,等.高效液相色谱法同时测定禽蛋中4种氟喹诺酮类药物残留量[J].分析仪器,2010(2):17-20.
[9]李娟.动物组织中11种喹诺酮类药物多残留的HPLC法研究[D].雅安:四川农业大学,2007.
[10]马建民,夏曦,李晓薇,等.阴离子交换固相萃取-超高效液相色谱-串联质谱法检测猪肌肉中13种喹诺酮类药物[J].中国食品卫生杂志,2013,25(3):249-253.
[11]刘柏林,谢继安,赵紫微,等.超高压液相色谱-电喷雾串联四极杆质谱内标法同时测定禽类食品中11种喹诺酮类药物[J].中国食品卫生杂志,2017,29(3):316-321.
[12]赵海香,孙艳红,丁明玉,等.多壁碳纳米管净化/超高效液相色谱串联质谱同时测定动物组织中四环素与喹诺酮多残留[J].分析测试学报,2011, 30(6):635-639.
[13]周鑫,李明,张鑫,等.高效液相色谱-串联质谱法测定鸡肉中的喹诺酮类和四环素类药物残留[J].畜牧与兽医,2015,47(11):19-22.
[14]倪永付,朱莉萍,王勇,等.微波辅助萃取/液相色谱-三重四极杆串联质谱法检测猪肉中氟喹诺酮类与四环素类药物残留[J].分析测试学报,2012,31(增刊):101-105.