 张昕,张晶,邵兵.固相萃取-超高效液相色谱-串联质谱法测定水中5种常用苯二氮卓类药物[J].中国食品卫生杂志,2017,29(6):684-689.
张昕,张晶,邵兵.固相萃取-超高效液相色谱-串联质谱法测定水中5种常用苯二氮卓类药物[J].中国食品卫生杂志,2017,29(6):684-689.DOi:10.13590/j.cjfh.2017.06.010
固相萃取-超高效液相色谱-串联质谱法测定水中5种常用 苯二氮卓类药物
(1.首都医科大学公共卫生学院,北京100069; 2.北京市疾病预防控制中心 食物中毒诊断溯源 技术北京市重点实验室,北京100013; 3.北京市预防医学研究中心,北京100013)
收稿日期:2017-08-22
作者简介:张昕女硕士生研究方向为食品安全检测E-mail:f_zhangxin@163.com
通信作者:邵兵男研究员研究方向为化学污染检测E-mail:shaobingch@sina.com
基金项目:国家自然科学基金(21677019);首都卫生发展科研专项项目(2014-1-3011)
摘要:目的建立超高效液相色谱-串联质谱(UPLC-MS/MS)检测方法,测定饮用水中硝西泮、地西泮、奥沙西泮、氯氮卓、氯硝西泮5种常用苯二氮卓类药物。方法500 ml水样经MCX柱富集净化,ACQUITY BEH C18(2.1 mm×100 mm,1.7 μm)色谱柱分离,以甲醇-水为流动相梯度洗脱,采用电喷雾正离子源(ESI+)在多反应监测模式(MRM)下进行测定,外标法定量。结果5种目标化合物在选定浓度范围内线性关系良好,相关系数(r2)均>0.999。方法的定量限(LOQ)为0.050~0.500 ng/L,3个添加水平的加标回收率为78.2%~108.0%,相对标准偏差≤12.4%(n=6)。利用该方法对96份饮用水样品进行分析,51份样品检出地西泮,浓度范围为 ~29.92>
关键词:
饮用水; 苯二氮卓类药物; 固相萃取; 超高效液相色谱-串联质谱法; 违法添加; 食品安全
文章编号:1004-8456(2017)06-0684-06
中图分类号:R155
文献标志码:A
Determination of five benzodiazepines in water by solid-phase extraction and ultra performance liquid chromatography-tandem mass spectrometry
( 1.School of Public Health, Capital Medical University, Beijing 100069, China; 2.Beijing Key Laboratory of Diagnostic and Traceability Technologies for Food Poisoning, Beijing Center for Disease Prevention and Control, Beijing 100013, China; 3.Beiji)
Abstract:ObjectiveA simultaneous determination method for five benzodiazepines (nitrazepam, diazepam, oxazepam, chlordiazepoxide, clonazepam) in drinking water was developed using ultra performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). MethodsSamples were concentrated and purified with MCX solid-phase extraction cartridge. The chromatographic separation was achieved using an ACQUITY BEH C18 (2.1 mm×100 mm, 1.7 μm) column with methanol and water as mobile phase. The targets were analyzed with ESI operating in positive multiple reaction monitoring (MRM) mode. Quantification was performed by matrix-matched standard calibration. ResultsSatisfactory linearity (r2>0.999) was obtained for all targets, with the limits of quantification (LOQs) ranged in 0.050-0.500 ng/L. Average recoveries at three spiked levels were 78.2%-108.0%, with relative standard deviations (RSDs) less than or equal to 12.4% (n=6). The developed method was used to analyze 96 drinking water samples, in which 51 samples were detected containing diazepam and 13 samples containing oxazepam. The concentrations were
Key words:
Drinking water; benzodiazepines; solid-phase extraction; ultra performance liquid chromatography-tandem mass spectrometry; adulterant; food safety
苯二氮卓类药物(benzodiazepines,BZDs)能够作用于大脑皮层中的γ-氨基丁酸(GABA)受体,抑制中枢神经系统活动,具有抗焦虑、抗惊厥及中枢性肌肉松弛作用[1],是临床常用的镇静剂[2],还可能被违法添加到动物饲料[3-4]和镇静类中成药[5]中。使用后的药物会随废水直接排放,或经人和动物代谢后经排泄进入生活污水处理系统或环境水体。这些水体通过蒸发、降雨、径流、渗透等途径污染地表水和地下水,给饮用水水源带来潜在威胁[6]。西班牙马德里地区的14份地表水样品中全部检出地西泮和劳拉西泮[7];欧洲七大主要流域的地表水样品(n=138)中,BZDs检出率高达86%[8];美国也曾在水源水样品中检测到浓度为0.47 ng/L的地西泮[9]。由于我国饮用水处理过程对这类痕量污染物的清除率极低[10],使得相关的药物可能在饮用水中存在。已有研究[11]表明,BZDs生物利用度高且半衰期长,能够穿透血脑屏障和血胎屏障对胎儿神经系统造成影响,并且药物在体内的累积对血液细胞、肾细胞也具有潜在的毒性作用[12],因此有必要对饮用水中的地西泮等药物进行关注。可靠的分析方法是开展样品监测和风险评估的基础,但现有的检测方法多集中于药物检测[4-5]、食品检测[3,13]、生物样本检测[14-15]以及环境水检测[7-8],对于饮用水中的检测则鲜有报道,因此有必要开展饮用水中多种BZDs的同时检测方法。
BZDs按其半衰期可分为短效类、中效类和长效类3种,目前临床使用较多的包括奥沙西泮(短效类)、氯氮卓(中效类)、地西泮(长效类)、硝西泮(长效类)和氯硝西泮(长效类)等。我国农业部第176号公告[16]明确规定禁止在饲料和动物饮用水中添加此类药物,国家食品药品监督管理总局制定的《保健食品中可能非法添加的物质名单(第一批)》[17]中,以上几种药物也被列入其中,因此,本试验将此5种常用BZDs作为研究对象,建立了水中BZDs超高效液相色谱-串联质谱(UPLC-MS/MS)的分析检测方法,并对某市96份生活饮用水样品中BZDs存在水平进行了初步评估。
BZDs标准物质硝西泮(CAS:146-22-5)、地西泮(CAS:439-14-5)、奥沙西泮(CAS:604-75-1)、氯氮卓(CAS:58-25-3)、氯硝西泮(CAS:1622-61-3),纯度均>99%,均购自中国食品药品检定研究院;甲醇、乙腈、氨水和超纯水均为质谱级,甲酸(>98%),乙酸铵(色谱级)。
质谱:离子源:电喷雾离子源(ESI+),毛细管电压2.8 kV,锥孔电压20 V,离子源温度150 ℃,脱溶剂气(N2)温度450 ℃,脱溶剂气流量1 000 L/h,碰撞气流速0.15 ml/min,多反应监测模式(MRM)采集数据,具体参数见表1。
400 ℃烘烤4 h以上。同时为了评价实验室的污染情况,每10份样品设定1个过程空白和1个质量控制点。
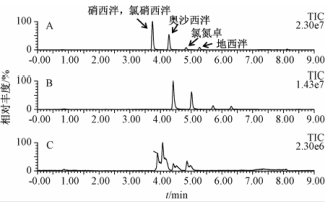
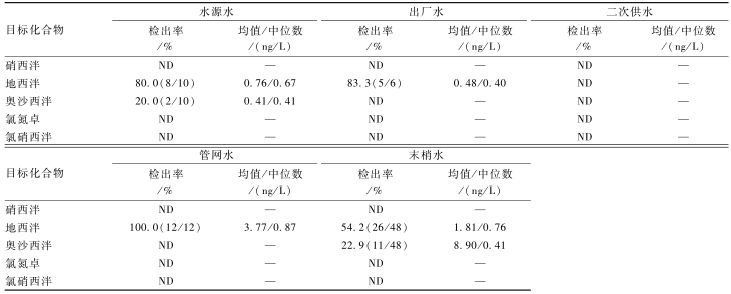
为85%)略有不同,这可能与不同地区药物使用情况的差异性有关。图3为混合标准品、过程空白和实际水样中目标化合物的MRM色谱图,样品C为某末梢水样品,其中地西泮和奥沙西泮的浓度分别为0.78和93.93 ng/L。以上结果说明,自来水厂现有的净化处理和消毒工艺并不能完全清除BZDs,饮用水中的这类药物应引起进一步关注。
BZDs按其半衰期可分为短效类、中效类和长效类3种,目前临床使用较多的包括奥沙西泮(短效类)、氯氮卓(中效类)、地西泮(长效类)、硝西泮(长效类)和氯硝西泮(长效类)等。我国农业部第176号公告[16]明确规定禁止在饲料和动物饮用水中添加此类药物,国家食品药品监督管理总局制定的《保健食品中可能非法添加的物质名单(第一批)》[17]中,以上几种药物也被列入其中,因此,本试验将此5种常用BZDs作为研究对象,建立了水中BZDs超高效液相色谱-串联质谱(UPLC-MS/MS)的分析检测方法,并对某市96份生活饮用水样品中BZDs存在水平进行了初步评估。
1材料与方法
1.1主要仪器与试剂
超高效液相色谱仪、XevoTM TQ-S三重四极杆质谱仪、Oasis HLB固相萃取柱(200 mg/6 ml)、Oasis MCX固相萃取柱(150 mg/6 ml)、C18固相萃取柱(200 mg/3 ml)、ACQUITY UPLC BEH C18(2.1 mm×100 mm,1.7 μm)、ACQUITY UPLC BEH Shield RP18(2.1 mm×100 mm,1.7 μm)、ACQUITY UPLC HSS T3(2.1 mm×100 mm,1.8 μm)色谱柱均购自美国Waters,PLEXA固相萃取柱(200 mg/6 ml,美国Agilent),Milli-Q超纯水机。BZDs标准物质硝西泮(CAS:146-22-5)、地西泮(CAS:439-14-5)、奥沙西泮(CAS:604-75-1)、氯氮卓(CAS:58-25-3)、氯硝西泮(CAS:1622-61-3),纯度均>99%,均购自中国食品药品检定研究院;甲醇、乙腈、氨水和超纯水均为质谱级,甲酸(>98%),乙酸铵(色谱级)。
1.2方法
1.2.1标准溶液的配制
分别准确称取10 mg标准品,溶解后转移至10 ml棕色容量瓶中,甲醇定容,配制成浓度为1 000 mg/L的标准储备液,保存于-20 ℃冰箱中。临用时用甲醇-水(50∶50,V/V)稀释标准溶液,配制成不同浓度的系列混合标准溶液(0.1、0.2、1、2、10、20、100、200 μg/L),分别取系列混合标准溶液1 ml复溶空白基质样品,得到基质匹配系列混合标准溶液。
1.2.2样品采集与预处理
2015年11月至2016年10月期间,采集某市3个区的水源水、出厂水、二次供水、管网水、末梢水样品共96份待测,每份1 L置于洁净的棕色玻璃瓶中。待运回实验室后立即经固相萃取柱富集净化。水样以5~10 ml/min的速度过MCX柱(分别用6 ml甲醇、6 ml超纯水活化),随后用6 ml超纯水,6 ml甲醇-水(40∶60,V/V)淋洗并抽干,最后用6 ml含3%氨水的乙腈洗脱。洗脱液在氮气流下吹至近干,用1 ml 甲醇-水(50∶50,V/V)定容,于4 ℃保存并于72 h内上机检测。
1.2.3仪器条件
色谱:色谱柱:ACQUITY UPLC BEH C18(2.1 mm×100 mm,1.7 μm),柱温40 ℃,进样体积10.0 μl。流动相A为甲醇,B为超纯水;流速0.3 ml/min;梯度洗脱条件:0~1 min 30%A,1~2 min 30%A~55%A,2~6 min 55%A~70%A,6~6.5 min 70%A~100%A,6.5~7 min保持100%A,7~7.1 min 100%A~30%A,7.1~9 min 30%A。质谱:离子源:电喷雾离子源(ESI+),毛细管电压2.8 kV,锥孔电压20 V,离子源温度150 ℃,脱溶剂气(N2)温度450 ℃,脱溶剂气流量1 000 L/h,碰撞气流速0.15 ml/min,多反应监测模式(MRM)采集数据,具体参数见表1。
1.2.4质量控制
试验中为了控制在样品采集与分析过程中可能存在的污染,所有玻璃容器均在马弗炉中
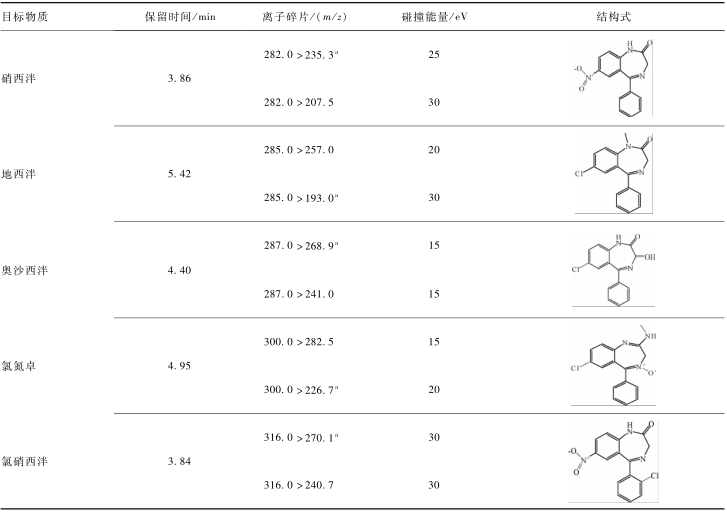
|
表1各目标化合物的结构式与质谱参数 Table 1Structure and MS/MS parameters for the target compounds 注:a为定量离子 |
2结果与分析
2.1液相条件的优化
2.1.1色谱柱的选择
本试验考察了BEH C18、HSS T3和BEH Shield RP18三种色谱柱对目标化合物色谱行为的影响。结果如图1所示,BEH C18柱能获得较好的分离效果,同时目标化合物的峰形更好,响应更高,基线噪音更低,因此本试验选择该色谱柱为分析柱。
|
注:A:BEH C18;B:HSS T3;C:BEH Shield RP18 图1不同色谱柱条件下的目标化合物色谱图 Figure 1Chromatograms of the target compounds by different UPLC columns |
2.1.2流动相条件的优化
本试验对不同流动相体系(甲醇-水、乙腈-水、甲醇-0.1%甲酸溶液、甲醇-5 mmol/L乙酸铵溶液、乙腈-0.1%甲酸溶液、乙腈-5 mmol/L乙酸铵溶液)进行考察。考虑到在ESI+模式下,酸性流动相体系可能会增强目标化合物的电离效率,改善峰形并获得更高的响应值,因此在水相中分别加入0.1%甲酸和5 mmol/L乙酸铵,然而结果显示两种体系并未产生预期的结果;并且当采用甲醇作为有机相时,与乙腈比较,目标化合物具有更高的灵敏度和更好的分离度,因此最终采用甲醇-水作为流动相进行试验。
2.2固相萃取条件的优化
2.2.1固相萃取柱的选择
由于饮用水样品中BZDs浓度很低,且基质中存在干扰,试验将采用固相萃取柱对样品进行富集净化。本试验选取了4种固相萃取柱HLB、MCX、PLEXA和C18做加标试验。预先用6 ml甲醇和6 ml超纯水活化萃取柱,选取500 ml空白水样品,加入100 μl 100 μg/L混合标准溶液,配制成加标浓度为20 ng/L的样品并振荡混匀,随后以5~10 ml/min 的流速过柱并经6 ml超纯水、6 ml甲醇-水(40∶60,V/V)淋洗抽干,随后用6 ml甲醇洗脱;MCX柱用6 ml超纯水、6 ml甲醇-水(40∶60,V/V)淋洗抽干后,用6 ml含3%氨水的乙腈洗脱。洗脱液在氮气流下吹至近干,甲醇-水(50∶50,V/V)定容至1 ml。每组试验重复3次,结果见表2。4种小柱对目标化合物的富集净化效果差异较大,HLB、PLEXA和C18柱的回收率分别为61.4%~79.6%、66.4%~83.3%和57.8%~76.2%,且基质效应较强,均不能满足试验要求;而经MCX柱处理后,目标化合物的回收率均>80%,基质效应降低到20%以下。这是由于BZDs多为极性较强的弱碱性化合物,而MCX柱是一种混合型阳离子交换固相萃取柱,对碱性化合物具有较高的选择性和灵敏度,因此最终选择MCX柱对样品进行前处理。
|
|
表2目标物质在不同固相萃取柱中的回收率基质效应结果(%) Table 2Recoveries and matrix effect using different solid phase extraction columns 注:回收率=(基质加标峰面积/基质匹配峰面积)×100%;基质效应=(1-基质匹配峰面积/标准品峰面积)×100% |
2.2.2上样液pH值的选择
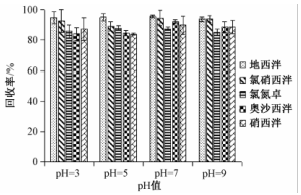
由于MCX柱具有阳离子交换和反相吸附两种模式,故当目标化合物离子化呈正电性时更易被吸附,在不同的pH值条件下,目标化合物具有不同的离子形态。调节上样液的pH值为3、5、7和9,分别进行加标回收试验。结果如图2所示,pH值的改变对目标化合物的回收率基本没有影响。原因是在几种pH值的条件下,目标化合物均能通过离子吸附作用较好的保留在MCX柱上。生活饮用水的pH值为6.5~8.5,因此无需调节样品pH值,直接过柱对样品进行富集净化。
|
图2目标化合物在不同上样液pH值条件下的回收率(n=6) Figure 2Recoveries of the target compounds in loading solution with different pH |
2.3线性范围、检出限与定量限
采用未检出目标化合物的饮用水样品配制系列混合标准溶液,以质量浓度x(μg/L)对应各目标化合物定量离子的峰面积y绘制基质匹配标准曲线,以3倍信噪比(S/N≥3)和10倍信噪比(S/N≥10)分别确定检出限(LOD)与定量限(LOQ)。方法的线性范围、LOD与LOQ如表3所示,结果表明,5种目标化合物在所选定的浓度范围内线性关系良好,相关系数(r2)均>0.999。地西泮和氯氮卓的LOQ均为0.050 ng/L,LOD均为0.025 ng/L;硝西泮、奥沙西泮和氯硝西泮的LOQ均为0.500 ng/L,LOD均为0.250 ng/L。
2.4方法回收率与精密度
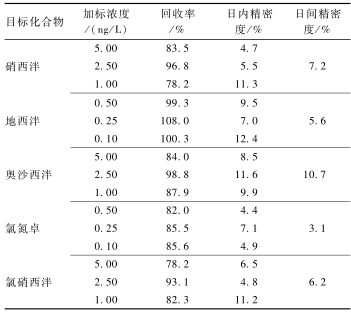
选取空白饮用水样品基质,加入低、中、高3个不同浓度水平的混合标准溶液,以考察方法的可靠性与稳定性,每个水平设6个平行,采用外标法计算各目标物质的回收率和相对标准偏差(RSD),同时选择高浓度连续进行5 d的加标回收试验,得到方法的日间精密度。结果如表4所示,方法的回收率为78.2%~108.0%,日内精密度为4.4%~12.4%,日间精密度为3.1%~10.7%,表明该方法具有良好的重现性与稳定性,可用于实际样品分析。
2.5实际样品检测
应用所建立的方法对96份饮用水样品进行定量分析,检测结果见表5。在51份样品中检出地西泮,浓度为<LOQ~29.92 ng/L,仅二次供水样品未检出;13份样品中检出奥沙西泮,浓度为<LOQ~93.93 ng/L,仅在水源水和末梢水中有检出;其余化合物在各类样品中均未检出。两种化合物的检出浓度均高于FICK等[8]报道的欧洲地表水中地西泮( |
|
表3方法的线性范围、LOD与LOQ Table 3Linearity, limits of detection and limits of quantification of the method |
|
表4饮用水样品不同浓度加标的回收率和相对标准偏差(n=6) Table 4Recoveries and relative standard deviations of the target compounds |
3小结
本试验采用MCX固相萃取柱富集净化,超高效液相色谱-串联质谱技术建立了饮用水中BZDs的检测方法。该方法前处理简便、线性良好、具有较高的重现性,可满足饮用水中BZDs的痕量检测要求,为相关部门开展风险监测提供了方法学依据。
|
表5饮用水样品检测结果(n=96) Table 5Occurrence of target compounds in drinking water samples 注:ND表示未检出;—表示无数据 |

|
注:A:标准溶液(各目标化合物浓度均为10 μg/L);B:过程空白;C:末梢水样品;CZP为氯硝西泮;CDP为氯氮卓;OZP为奥沙西泮;DZP为地西泮;NZP为硝西泮 图3目标化合物的MRM色谱图 Figure 3Multiple reaction monitoring (MRM) chromatograms of analytes in different samples |
参考文献
[1]FERNNDEZ P, GONZLEZ C, PENA M T, et al. A rapid ultrasound-assisted dispersive liquid-liquid microextraction followed by ultra-performance liquid chromatography for the simultaneous determination of seven benzodiazepines in human plasma samples[J]. Analytica Chimica Acta, 2013, 767(5): 88-96.
[2]OCHOA J G, KILGO W A. The role of benzodiazepines in the treatment of epilepsy[J]. Current Treatment Options in Neurology, 2016, 18(4): 1-11.
[3]YAN L J, ZHANG F, MIN W U, et al. Determination of benzodiazepines in animal feeds by UPLC-ESI MS/MS[J]. Journal of Instrumental Analysis, 2011, 30(11): 1301-1305.
[4]贾玉香, 朱若华. HPLC法快速测定饲料中地西泮的含量[J]. 分析化学, 2009, 37(增刊): 207.
[5]朱琳, 阮丽萍, 刘华良,等. 高效液相色谱-串联质谱法测定保健食品中的23种精神药品[J]. 色谱, 2013, 31(7): 709-713.
[6]GARCA-GALN M J, DAZ-CRUZ M S, BARCEL D. Identification and determination of metabolites and degradation products of sulfonamide antibiotics[J]. Trac Trends in Analytical Chemistry, 2008, 27(11): 1008-1022.
[7]MENDOZA A, LPEZ D A M, GONZLEZALONSO S, et al. Occurrence of drugs of abuse and benzodiazepines in river waters from the Madrid Region (Central Spain)[J]. Chemosphere, 2014, 95(1): 247-255.
[8]FICK J, BRODIN T, HEYNEN M, et al. Screening of benzodiazepines in thirty European rivers[J]. Chemosphere, 2017, 176(2): 324-332.
[9]GERRITY D W, BENOTTI M J, RECKHOW D A, et al. Pharmaceuticals and endocrine-disrupting compounds in drinking water[J]. Environmental Science & Technology, 2011, 43(3): 597-603.
[10]白晓慧, 贺兰喜, 王宝贞. 常规饮用水净化技术面临的挑战及对策[J]. 水科学进展, 2002, 13(1): 122-127.
[11]ODSBU I, SKURTVEIT S, SELMER R, et al. Prenatal exposure to anxiolytics and hypnotics and language competence at 3 years of age.[J]. European Journal of Clinical Pharmacology, 2015, 71(3): 283-291.
[12]李想, 蒋可心. 饮用水中苯二氮卓类药物残留研究现状[J]. 中国初级卫生保健, 2010, 24(7): 60-62.
[13]严爱花, 李贤良, 郗存显,等. 固相萃取-液相色谱-串联质谱法同时检测猪肉中18种苯二氮卓类药物残留量[J]. 食品与发酵工业, 2013, 39(4): 173-179.
[14]RAHIMI-NASRABADI M, KHOSHROO A, MAZLOUM-ARDAKANI M. Electrochemical determination of diazepam in real samples based on fullerene-functionalized carbon nanotubes/ionic liquid nanocomposite[J]. Sensors & Actuators B Chemical, 2017, 240(8): 125-131.
[15]KIM J, LEE S, IN S, et al. Validation of a simultaneous analytical method for the detection of 27 benzodiazepines and metabolites and zolpidem in hair using LC-MS/MS and its application to human and rat hair[J]. Journal of Chromatography B, 2011, 879(13/14): 878-886.
[16]中华人民共和国农业部. 中华人民共和国农业部公告第176号[A/OL]. (2011-04-22)[2017-07-15]. http://www.moa.gov.cn/zwllm/tzgg/gg/201104/t20110422_1976307.htm.
[17]国家食品药品监督管理总局. 保健食品中可能非法添加的物质名单(第一批)[A/OL]. (2012-12-14)[2017-07-15]. http://www.sda.gov.cn/WS01/CL1162/76614.html.
[2]OCHOA J G, KILGO W A. The role of benzodiazepines in the treatment of epilepsy[J]. Current Treatment Options in Neurology, 2016, 18(4): 1-11.
[3]YAN L J, ZHANG F, MIN W U, et al. Determination of benzodiazepines in animal feeds by UPLC-ESI MS/MS[J]. Journal of Instrumental Analysis, 2011, 30(11): 1301-1305.
[4]贾玉香, 朱若华. HPLC法快速测定饲料中地西泮的含量[J]. 分析化学, 2009, 37(增刊): 207.
[5]朱琳, 阮丽萍, 刘华良,等. 高效液相色谱-串联质谱法测定保健食品中的23种精神药品[J]. 色谱, 2013, 31(7): 709-713.
[6]GARCA-GALN M J, DAZ-CRUZ M S, BARCEL D. Identification and determination of metabolites and degradation products of sulfonamide antibiotics[J]. Trac Trends in Analytical Chemistry, 2008, 27(11): 1008-1022.
[7]MENDOZA A, LPEZ D A M, GONZLEZALONSO S, et al. Occurrence of drugs of abuse and benzodiazepines in river waters from the Madrid Region (Central Spain)[J]. Chemosphere, 2014, 95(1): 247-255.
[8]FICK J, BRODIN T, HEYNEN M, et al. Screening of benzodiazepines in thirty European rivers[J]. Chemosphere, 2017, 176(2): 324-332.
[9]GERRITY D W, BENOTTI M J, RECKHOW D A, et al. Pharmaceuticals and endocrine-disrupting compounds in drinking water[J]. Environmental Science & Technology, 2011, 43(3): 597-603.
[10]白晓慧, 贺兰喜, 王宝贞. 常规饮用水净化技术面临的挑战及对策[J]. 水科学进展, 2002, 13(1): 122-127.
[11]ODSBU I, SKURTVEIT S, SELMER R, et al. Prenatal exposure to anxiolytics and hypnotics and language competence at 3 years of age.[J]. European Journal of Clinical Pharmacology, 2015, 71(3): 283-291.
[12]李想, 蒋可心. 饮用水中苯二氮卓类药物残留研究现状[J]. 中国初级卫生保健, 2010, 24(7): 60-62.
[13]严爱花, 李贤良, 郗存显,等. 固相萃取-液相色谱-串联质谱法同时检测猪肉中18种苯二氮卓类药物残留量[J]. 食品与发酵工业, 2013, 39(4): 173-179.
[14]RAHIMI-NASRABADI M, KHOSHROO A, MAZLOUM-ARDAKANI M. Electrochemical determination of diazepam in real samples based on fullerene-functionalized carbon nanotubes/ionic liquid nanocomposite[J]. Sensors & Actuators B Chemical, 2017, 240(8): 125-131.
[15]KIM J, LEE S, IN S, et al. Validation of a simultaneous analytical method for the detection of 27 benzodiazepines and metabolites and zolpidem in hair using LC-MS/MS and its application to human and rat hair[J]. Journal of Chromatography B, 2011, 879(13/14): 878-886.
[16]中华人民共和国农业部. 中华人民共和国农业部公告第176号[A/OL]. (2011-04-22)[2017-07-15]. http://www.moa.gov.cn/zwllm/tzgg/gg/201104/t20110422_1976307.htm.
[17]国家食品药品监督管理总局. 保健食品中可能非法添加的物质名单(第一批)[A/OL]. (2012-12-14)[2017-07-15]. http://www.sda.gov.cn/WS01/CL1162/76614.html.