高效液相色谱-三重四级杆质谱法同时测定水产品中6种苯二氮卓类镇静剂残留
张 律[1]*, 朱 波, 赖少阳, 岳亚军, 游杰
(深圳市罗湖区疾病预防控制中心, 广东深圳 518020)
摘 要: 目的 建立高效液相色谱-三重四级杆质谱法同时测定水产品中6种苯二氮卓类镇静剂残留的方法。方法 样品用乙腈提取后经C18固相萃取柱净化, 待测液经Waters ACQUITY UPLC BEH C18色谱柱(2.1 mm×50 mm, 1.7 μm)分离, 以0.1%甲酸水溶液和甲醇为流动相进行梯度洗脱, 采用多反应监测模式检测, 内标法定量。 结果 6种苯二氮卓类镇静剂在1.0~50.0 μg/L浓度范围内线性关系良好, 相关系数r均>0.999, 检出限和定量限分别为0.3 μg/kg和1.0 μg/kg, 在3个浓度加标水平下的平均回收率为74.2%~108.0%, 相对标准偏差为1.1%~6.7%。 结论 该方法简便快捷, 灵敏度高, 结果准确, 可用于水产品中6种苯二氮卓类镇静剂残留的同时检测。
关键词: 高效液相色谱-三重四级杆质谱法;水产品;苯二氮卓;镇静剂
Simultaneous determination of six benzodiazepine sedatives residue in aquatic products by high performance liquid chromatography-triple quadrupole mass spectrometry
ZHANG Lv*,ZHU Bo,LAI Shao-yang,YUE Ya-jun,YOU Jie
( Luohu Center for Disease Control and Prevention,Shenzhen 518020,China)
Abstract: Objective To establish a method for the simultaneous determination of 6 benzodiazepine sedatives residue in aquatic products by high performance liquid chromatography-triple quadrupole mass spectrometry. Methods The samples were extracted with acetonitrile and purified by C18 solid phase extraction column. The sample solution was separated by Waters ACQUITY UPLC BEH C18 column(2.1 mm×50 mm, 1.7 μm) using 0.1% formic acid and methanol as mobile phase for gradient elution, determined in multiple reaction monitoring mode and quantified by internal standard method. Results 6 benzodiazepine sedatives had a good linear relationship in the range of 1.0~50.0 μg/L with r>0.999, the limits of detection and limits of quantification were 0.3 μg/kg and 1.0 μg/kg. Average recoveries for the analytes at 3 spiked levels ranged from 74.2%~108.0% with relative standard deviations of 1.1%~6.7%. Conclusion The method is simple, rapid, sensitive and accurate, which is suitable for simultaneous determination of 6 benzodiazepine sedatives residue in aquatic products.
Keywords: high performance liquid chromatography-triple quadrupole mass spectrometry;aquatic products; benzodiazepine;sedative
苯二氮卓类镇静剂(BZDs)是临床上常用的一类安定药物,能够与大脑皮层中的γ-氨基丁酸(GABA)受体结合,从而起到神经抑制的效果。因具有抗焦虑、抗惊厥、松弛肌肉、镇痛、镇静催眠等作用[1]被广泛应用于治疗失眠、抑郁、癫痫等疾病上。主要包括地西泮、奥沙西泮、硝西泮、氯硝西洋、去甲西泮、劳垃西泮、替马西泮、氟硝西泮等。近年来,一些不法商家擅自在鲜活水产捕捞和运输过程中非法使用镇静剂,以减少鱼体碰撞造成的伤害,从而降低运输途中的死亡率[2-3]。此类药物由于代谢缓慢,极易在动物组织中蓄积残留,人体长期摄入后易引起嗜睡疲乏、记忆退化、运动障碍等一系列问题[4]。据文献统计,2018年以来,天津、上海、江苏、浙江、湖北等地均有水产品中地西泮残留事件的报道 [5-7]。GB 31650-2019《食品安全国家标准 食品中兽药最大残留限量》中也有明确规定:不得在动物性食品中检出地西泮、氯丙嗪等药物残留,该类物质仅允许在治疗途径上使用。
目前,国内外镇静剂检测的方法主要有酶联免疫法[8]、气相色谱法、气相色谱-质谱联用法、高效液相色谱法[9]、高效液相色谱-三重四级杆质谱法[10-11]等。其中,酶联免疫法稳定性较差,易出现假阳性,需要用其他方法进一步确证;气相色谱-质谱联用法需衍生处理,操作繁琐,不适合检测分析大批量样品;气相色谱法、高效液相色谱法仅以保留时间定性,灵敏度较低,且易受复杂基质干扰,不适合药物残留的痕量分析;高效液相色谱-三重四级杆质谱法因灵敏度高、定性定量准确、不需要衍生等优点,成为目前镇静剂类药物残留检测的重要检测手段[12]。因水产品中含有丰富的蛋白质和脂肪,样品前处理方法若不能有效地消除基质效应,将对检测的灵敏度和回收率造成严重影响。现有研究有采用液液萃取、正己烷除脂等方法进行净化[13-14],在去除杂质干扰的同时易造成目标物损失。国内还有学者在前处理过程中采用QuEChERS方法进行净化[15],操作较繁琐,净化效果较差。
本研究通过对仪器分析条件及前处理方式的改进和优化,建立了C18固相萃取柱净化样品,同位素内标法定量,高效液相色谱-三重四级杆质谱技术同时测定水产品中地西泮、奥沙西泮、硝西泮、氯硝西泮、劳拉西泮、去甲西泮等6种苯二氮卓类镇静剂药物残留的检测方法,以期为水产品中苯二氮卓类镇静剂药物残留的定性定量分析提供技术参考。
1 材料与方法
1.1 主要仪器与试剂
高效液相色谱-三重四级杆质谱仪,带电喷雾离子源(ESI)(AB SCIEX 5500+,美国 AB SCIEX 公司);T25组织匀浆机,VX-200漩涡混合机,HS501温控摇床(德国IKA公司);BSA623S型电子天平(赛多利斯北京有限公司);KQ-600DE数控超声波清洗器(昆山市超声仪器有限公司);SIGMA 3K30台式高速冷冻离心机(德国SIGMA公司);TorbovapLV多功能全自动样品浓缩仪(英国Biotage公司);Supelco SPE-24管手动固相萃取装置(美国Supelco公司);Milli-Q IQ7000超纯水仪(法国Millipore公司)。
CNWBOND HC- C18固相萃取柱(500 mg/3 mL,上海安谱实验科技股份有限公司) ; SampliQ C18 ODS(500 mg/3 mL,美国Agilent公司);Oasis HLB固相萃取柱(60 mg/3cc);Oasis MCX固相萃取柱(60 mg/3cc)均购自美国Waters公司;地西泮(1 mg/mL)、奥沙西泮(1 mg/mL)、氯硝西泮(1 mg/mL)、去甲西泮(1 mg/mL)、硝西泮(1 mg/mL)、劳拉西泮(1 mg/mL)、地西泮- D5(100 μg/mL)、奥沙西泮-D5(100 μg/mL)、劳拉西泮-D4(100 μg/mL)均购自美国Cerilliant公司;甲醇、乙腈、甲酸为色谱纯;无水硫酸钠、无水硫酸镁、氯化钠为分析纯;试验用水为超纯水。
1.2 方法
1.2.1 标准溶液配制
单标准储备溶液的配制:用50%乙腈分别稀释标准物质溶液及同位素内标溶液至质量浓度为10 mg/L和1 mg/L, 于-20℃下避光保存。
混合标准使用液的配制: 将6种单标标准储备液用50%乙腈稀释成质量浓度为1000 μg/L的混合标准溶液, 4℃避光保存。
混合内标使用液的配制: 将4种单标内标储备液用50%乙腈稀释成质量浓度为200 μg/L的混合内标溶液, 4℃避光保存。
1.2.2 样品前处理
称取2.0 g充分匀质的样品于50 mL离心管中,准确加入50 μL混合内标使用液,再加入10 mL乙腈,涡旋混匀30 min后置于摇床上振荡30 min,超声提取20 min。加入1 g氯化钠和4 g无水硫酸钠,在4˚C条件下以8000 r/min的速度离心5 min,上清液全部转移至另一个洁净的离心管中,残渣再用5 mL乙腈溶液重复提取一次。合并上清液,旋涡混匀,于40˚C氮吹至干,再用40%甲醇3 mL复溶,在4˚C条件下以8000 r/min的速度离心5 min后待净化。C18固相萃取柱依次用3 mL甲醇、3 mL水活化,提取液以2~3mL/min的速度过柱后,用40%甲醇3 mL淋洗,抽干柱内液体后加入3 mL甲醇洗脱,收集洗脱液于40˚C氮吹至干,再用初始流动相定容至1 mL刻度处,经0.22 μm有机相滤膜过滤后待测定。
1.3 仪器条件
1.3.1 质谱条件
电喷雾离子源正离子(ESI+)模式;多反应监测(MRM)模式;离子化电压5500 V;离子源温度500℃;气帘气压力276kPa,喷雾气压力345kPa,辅助加热气压力345kPa;喷撞气压力模式为中模式。其他质谱参数见表1。
表1 6种镇静剂和4种同位素内标的质谱参数
Table 1 MS parameters of 6 sedatives and 4 isotope internal standards
化合物 | 保留时间(min) | 母离子(m/z) | 子离子(m/z) | 去簇电压(V) | 碰撞能量(eV) | 内标 | |||
地西泮 | 3.12 | 285.1 | 193.1*/154.1 | 136 | 44/38 | 地西泮- D5 | |||
奥沙西泮 | 2.10 | 287.0 | 241.0*/269.2 | 112 | 30/22 | 奥沙西泮-D5 | |||
硝西泮 | 2.08 | 282.1 | 236.1*/180.2 | 110 | 34/44 | 奥沙西泮-D5 | |||
氯硝西泮 | 2.22 | 316.1 | 270.1*/214.0 | 132 | 34/43 | 奥沙西泮-D5 | |||
劳拉西泮 | 2.23 | 321.1 | 275.1*/229.1 | 110 | 28/39 | 劳拉西泮-D4 | |||
去甲西泮 | 2.65 | 271.0 | 140.0*/165.2 | 120 | 38/40 | 去甲西泮-D5 | |||
地西泮- D5 | 3.09 | 289.9 | 198.2 | 120 | 40 | ||||
奥沙西泮-D5 | 2.07 | 292.0 | 246.1 | 100 | 32 | ||||
劳拉西泮-D4 | 2.21 | 325.0 | 279.0 | 110 | 30 | ||||
去甲西泮-D5 | 2.62 | 276.0 | 213.1 | 120 | 40 | ||||
注:*为定量离子
1.3.2 色谱条件
色谱柱:Waters ACQUITY UPLC BEH C18 (2.1 mm×50 mm,1.7 μm) ;柱温:45℃;进样体积:2 μL;流动相A:0.1%甲酸水溶液,流动相B:甲醇;流速:0.3 mL/min。梯度洗脱程序见表2。
表2 梯度洗脱程序
Table 2 Gradient elution program
时间(min) | A(%) | B(%) |
0.00 | 70 | 30 |
0.50 | 70 | 30 |
4.00 | 30 | 70 |
4.50 | 5 | 95 |
5.00 | 5 | 95 |
5.10 | 70 | 30 |
7.00 | 70 | 30 |
2 结果
2.1 前处理条件的选择
2.1.1 提取溶剂的选择
苯二氮卓类镇静剂为脂溶性物质,在常用的有机溶剂中均有溶解性。文献中常用的提取溶剂有甲醇、乙腈、正己烷、乙酸乙酯、氯仿、二氯甲烷[13]等。有文献报道,正己烷作为提取溶剂时,对地西泮具有较大的吸附作用从而易造成目标物损失,且提取液含有较多油脂等杂质,给后续净化带来一定困难[12];氯仿、二氯甲烷比重较大,提取和离心后样品悬浮于提取溶剂上方,致使提取液较难分离,操作不便[13];乙酸乙脂作为提取溶剂时,样品提取液乳化现象严重,回收率偏低[3]。试验以5 μg/kg加标鲩鱼样品为例,分别比较了甲醇、乙腈、0.1%(体积分数)甲酸乙腈作为提取溶剂时,6种苯二氮卓类镇静剂的回收率。试验发现,经甲醇提取后样液较浑浊,在后续净化过程中极易造成固相萃取柱的堵塞,且甲醇极性较强,提取出来的杂质较多,给分析带来一定干扰。如图1所示,使用甲醇提取时6种镇静剂的回收率均较乙腈和0.1%甲酸乙腈低。采用0.1%甲酸乙腈作为提取溶剂时地西泮、奥沙西泮、硝西泮和去甲西泮的回收率与纯乙腈的回收率无明显差异,氯硝西泮的回收率较纯乙腈低。综合考虑,后续试验选择乙腈作为提取溶剂。
![]()
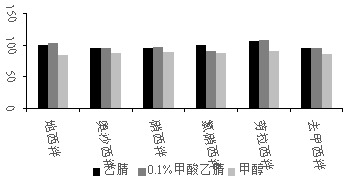
图1 不同提取溶剂对鱼肉中6种镇静剂回收率的影响
Figure 1 Effects of different extraction solvents on the recoveries of 6 sedatives in fish samples
2.1.2 盐析剂及提取次数的选择
鱼肉中的水分通常会影响后续的氮吹浓缩过程,前处理中加入盐析剂有利于待测物萃入有机相。试验以5 μg/kg加标鲩鱼样品为例,10 mL乙腈为提取溶剂,分别对比了添加1 g氯化钠和1 g无水硫酸镁、添加1 g氯化钠和4 g无水硫酸钠、乙腈提取1次以及提取2次对6种镇静剂回收率的影响。由于前期实验添加1 g氯化钠和4 g无水硫酸镁时仅收集到5 mL乙腈提取液, 且体系放热, 因此减少无水硫酸镁用量为1 g。如图2所示:添加1 g氯化钠和4 g无水硫酸钠,提取次数为2次时各目标物可获得最高回收率。与此同时考虑到无水硫酸镁溶于水后易产生较大热量,造成目标物损失,因此,选择1 g氯化钠和4 g无水硫酸钠作为盐析剂,提取次数为2次。为提高浓缩效率,第2次提取时乙腈的添加量为5 mL。
![]()
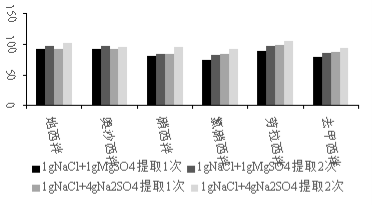
图2 不同盐析剂及提取次数对鱼肉中6种镇静剂回收率的影响
Figure 2 Effects of different salting-out agents and extraction time on the recoveries of 6 sedatives in fish samples
2.1.3 固相萃取柱的选择
针对苯二氮卓类化合物的性质,试验研究了HC-C18、SampliQ C18 ODS、Oasis HLB、Oasis MCX 4种不同固相萃取柱的净化效果。结果显示:SampliQ C18 ODS柱对于奥沙西泮、硝西泮、氯硝西泮、劳拉西泮的保留性较差,回收率不理想。HC-C18柱对6种苯二氮卓类镇静剂均有较高的回收率,见图3。故最终选择HC-C18固相萃取柱进行净化。
![]()
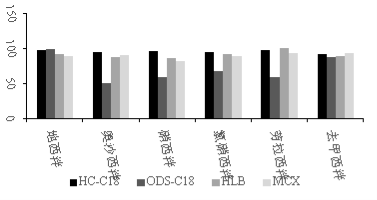
图3 不同固相萃取小柱对鱼肉中6种镇静剂回收率的影响
Figure 3 Effects of different solid phase extraction column on the recoveries of 6 sedatives in fish samples
2.1.4 复溶液、淋洗液以及洗脱液的选择
分别收集30%、40%和50%甲醇溶液配制的复溶液过柱后进行测定,发现用50%甲醇溶液配制的复溶液过柱后有检出微量奥沙西泮、硝西泮及氯硝西泮。样品上柱后,部分杂质会随被测物一起被吸附在固相萃取柱上,需淋洗去除杂质。收集30%、40%和50%甲醇溶液配制的淋洗液过柱后进行测定,发现50%甲醇溶液配制的淋洗液中检出奥沙西泮、硝西泮、氯硝西泮及劳拉西泮,说明50%甲醇溶液配制的复溶液和淋洗液对小柱有穿透作用,容易造成目标物损失。体积分数为40%的甲醇淋洗液既能有效去除杂质干扰,又不会将目标化合物洗脱。综合考虑,选择40%甲醇溶液作为复溶液和淋洗液。试验以纯甲醇作为洗脱液,2 mL的用量即可完全洗脱6种目标物。
2.2 流动相的选择
正离子电离模式下,在流动相中加入甲酸,有助于待测物质的离子化,从而提高目标化合物的响应值和分离度。苯二氮卓类镇静剂为极性化合物,适合采用反相色谱柱分离。试验采用ACQUITY UPLC BEH C18柱(2.1 mm×50 mm,1.7 μm)为固定相,分别比较了0.1%甲酸-甲醇和0.1%甲酸-乙腈两种流动相体系对6种苯二氮卓类镇静剂的分离效果的影响。结果显示:相比于0.1%甲酸-乙腈体系,0.1%甲酸-甲醇体系中的目标化合物具有更高的响应值和更低的背景噪音,峰型更对称,分离效果更好。因此,试验流动相选择0.1%甲酸-甲醇体系。在优化条件下,空白皖鱼样品、5 μg /kg空白皖鱼加标样品及10 μg/L镇静剂混合标准溶液的总离子流色谱图见图4。
![]()
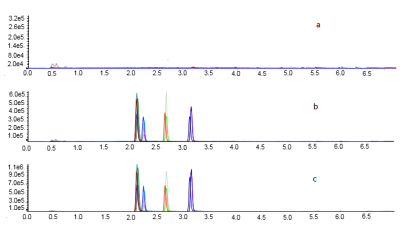
时间/time
图4 不同样品的总离子流色谱图
Figure 4 Total ion chromatograms of different samples
注:a为空白皖鱼样品;b为5 μg/kg-1空白皖鱼加标样品;c为10 μg/L镇静剂混合标准溶液
2.3 方法学试验结果
2.3.1 线性范围和检出限
吸取适量的混合标准溶液和混合内标溶液,用30%甲醇水稀释成1.0、2.0、5.0、10.0、20.0、50.0 μg/L系列标准溶液,每个浓度标准溶液添加混合内标溶液50 μL。以目标化合物的浓度为横坐标(X),以目标化合物与其同位素内标的峰面积比为纵坐标(Y),绘制标准曲线。结果表明:6种镇静剂在1.0~50.0 μg/L范围内线性关系良好,相关系数r均大于0.999。以空白加标样品3倍和10倍信噪比对应的浓度确定方法的检出限(LOD)和定量限(LOQ),结果见表3。
表3 6种镇静剂的线性范围、检出限(LOD)、定量限(LOQ)
Table 3 Linear ranges, limis of detection and limits of quantification for 6 sedatives
化合物 | 线性范围(μg/L) | 回归方程 | 相关系数r | LOD(μg/kg) | LOQ(μg/kg) |
地西泮 | 1.0~50.0 | Y=0.13327X-0.01385 | 0.9994 | 0.3 0.3 0.3 0.3 0.3 0.3 | 1.0 1.0 1.0 1.0 1.0 1.0 |
奥沙西泮 | 1.0~50.0 | Y=0.10699X-0.01145 | 0.9997 | ||
硝西泮 | 1.0~50.0 | Y=0.12894X-0.02983 | 0.9996 | ||
氯硝西泮 劳拉西泮 去甲西泮 | 1.0~50.0 1.0~50.0 1.0~50.0 | Y=0.04816X-0.00570 Y=0.07800X+0.01040 Y=0.17882X-0.02447 | 0.9995 0.9998 0.9996 |
2.3.2 回收率和精密度
分别选取皖鱼、基围虾、青蟹3种基质空白样品进行低、中、高3个浓度水平的加标回收试验,每个浓度水平重复测定6次,计算回收率和测定值的相对标准偏差(RSD)。如表4所示:6种镇静剂的回收率为74.2%~108.0%,测定值的RSD为1.1%~6.7%。
表4 鱼、虾、蟹样品中6种镇静剂的加标回收率及相对标准偏差(n=6)
Table 4 Recoveries and RSDs of 6 sedatives spiked in the fish, shrimp and crab samples(n=6)
化合物 | 加标浓度(μg/kg) | 皖鱼 | 基围虾 | 青蟹 | ||||
回收率(%) | RSD(%) | 回收率(%) | RSD(%) | 回收率(%) | RSD(%) | |||
地西泮 | 1 | 108.0 | 3.7 | 96.5 | 2.7 | 105.5 | 5.0 | |
2.5 | 97.6 | 2.8 | 93.0 | 2.4 | 101.7 | 4.3 | ||
5 | 100.2 | 3.5 | 92.2 | 2.1 | 103.1 | 2.9 | ||
奥沙西泮 | 1 | 99.0 | 3.9 | 101.5 | 4.5 | 100.6 | 5.0 | |
2.5 | 94.9 | 1.1 | 98.2 | 4.0 | 99.6 | 2.2 | ||
5 | 95.4 | 2.3 | 97.9 | 2.3 | 98.0 | 2.4 | ||
硝西泮 | 1 | 103.8 | 3.1 | 84.9 | 4.7 | 90.4 | 4.8 | |
2.5 | 97.4 | 3.0 | 77.5 | 4.5 | 87.3 | 4.4 | ||
5 | 95.6 | 4.2 | 74.2 | 1.1 | 88.2 | 3.9 | ||
氯硝西泮 | 1 | 103.7 | 4.5 | 106 | 6.1 | 104.9 | 3.9 | |
2.5 | 97.9 | 3.7 | 98.9 | 3.0 | 99.8 | 1.7 | ||
5 | 99.6 | 3.8 | 101.0 | 3.1 | 99.2 | 1.3 | ||
劳拉西泮 | 1 | 96.7 | 6.7 | 103.1 | 4.0 | 96.7 | 6.1 | |
2.5 | 106.2 | 2.0 | 99.8 | 4.6 | 99.9 | 2.1 | ||
5 | 106.3 | 3.5 | 96.6 | 3.5 | 98.5 | 3.8 | ||
去甲西泮 | 1 | 97.0 | 2.6 | 97.2 | 2.9 | 100.6 | 3.4 | |
2.5 | 90.5 | 3.8 | 96.0 | 2.5 | 99.8 | 2.4 | ||
5 | 94.4 | 4.6 | 95.1 | 4.9 | 96.2 | 5.2 | ||
2.4 实际样品检测
利用建立的方法对随机采自深圳市农贸市场和超市的60份水产品(包括乌鳢、鳊鱼、鲈鱼、草鱼、罗氏虾、大头虾、淡水虾、大闸蟹、膏蟹等)进行苯二氮卓类镇静剂残留筛查, 均未检出上述6种苯二氮卓类镇静剂药物残留。
3 结论
本研究建立了C18固相萃取柱净化样品,内标法定量,高效液相色谱-三重四级杆质谱法同时测定水产品中6种苯二氮卓类镇静剂药物残留的方法。该方法操作简便、灵敏度高、基质效应低、定性定量准确,可实现快速分析大批量样品,为水产品中多种苯二氮卓类镇静剂药物残留的同时测定及食品安全监管提供技术参考。
参考文献
[1] 严爱花,李贤良,郗存显,等.固相萃取-液相色谱-串联质谱法同时检测猪肉中18种苯二氮卓类药物残留量[J].食品与发酵工业,2013,39(4):173-179.
[1] YAN AH, LI XL, XI CX, et al. Simultaneous determination of 18 benzodiazepines in pork by solid phase extraction and liquid chromatography-tandem mass spectrometry[J]. Food Fermentation Industries, 2013, 39(4) : 173-179.
[2] 杨霄,万译文,黄华伟,等.分散固相萃取-超高效液相色谱-串联质谱法测定水产品中5种硝基咪唑类和6种苯二氮卓类药物[J].色谱,2022,40(7):625-633.
[2] YANG X, WAN YW, HUANG HW, et al.Determination of five nitroimidazoles and six benzodiazepines in aquatic products using ultra-high performance liquid chromatography-tandem mass spectrometry coupled with dispersive solid-phase extraction[J]. Chinese Journal of Chromatography, 2022, 40(7) : 625-633.
[3] 宿书芳,孙立臻,薛霞,等.通过式固相萃取-超高效液相色谱-串联质谱法测定水产品中地西泮[J].色谱,2020,38(7):791-797.
[3] SU SF, SUN LZ, XUE X, et al.Determination of diazepam in aquatic products by ultra performance liquid chromatography-tandem mass spectrometry with pass-through solid phase extraction[J]. Chinese Journal of Chromatography, 2020, 38(7) : 791-797.
[4] 张秋云,杨洪生,谭秀慧,等.液相色谱-串联质谱测定水产品中15种苯二氮卓类镇静剂的药物残留[J]. 食品与机械,2022,38(11):60-67,94.
[4] ZHANG QY, YANG HS, TAN XH, et al.Determination of the residues of 15 kinds of benzodiazepine sedatives in aquatic products by liquid chromatography tandem mass spectrometry[J]. Food and Machinery, 2022, 38(11) : 60- 67, 94.
[5] 王守英,孔聪,杨光昕,等.上海市售水产品地西泮及去甲地西泮残留调查分析[J].农产品质量与安全,2020(3):31-35,67.
[5] WANG SY, KONG C, YANG GX, et al. Diazepam and nordazepam residue in the aquatic product from market in Shanghai [J]. Quality and Safety of Agricultural products, 2020(3): 31-35, 67.
[6] 殷耀,张晓燕,于思然,等.江苏省市售淡水养殖鱼药物残留状况及分析[J].水产研究,2018,5(2):30-36.
[6] YIN Y, ZHANG XY, YU SR, et al. Analysis of drug residues in freshwater fishes in market of Jiangsu Province[J]. Open Journal of Fisherles Research, 2018, 5(2): 30-36.
[7] 纪律,刘晓俊,万正杨,等.2021年湖北省4个城市市售淡水鱼中兽药及非法添加残留监测结果分析[J]. 职业与健康,2022,38(23):3292-3295.
[7] JI L, LIU XJ, WAN ZY, et al. Analysis on motoring results of veterinary drugs and illegal additives residues in commercially available freshwater fish in four cities of Hubei Province in 2021[J].Occupation and Health, 2022, 38(23):3292-3295.
[8] 柯浩堃,吕赛男,郝红霞,等.苯二氮卓类化合物的检测方法研究进展[J].分析测试室,2020,39(9):1110-1116.
[8] KE HK, LUY SN, HAO HX, et al. Advances in the detection of benzodiazepines[J]. Chinese Journal of Analysis Laboratory, 2020, 39(9): 1110-1116.
[9] SRUTHI A, TEJASWI P, THANUJA N, et al.Simple RP-HPLC method for estimation of diazepam in tablet dosage form[J].Journal of Pharmacy Research, 2013, 6(1): 140-144.
[10] ORFANIDIS A, GIKA H, MASTROGIANNI O, et al.Determination of drugs of abuse and pharmaceuticals in skeletal tissue by UHPLC-MS/MS[J].Forensic Science International, 2018, 290: 137-145.
[11] 李芹,穆树荷,韩刚,等.水产品中镇静剂残留检测技术研究进展[J].中国农学通报,2021,37(12):86-91.
[11] LI Q, MU SH, HAN G, et al.Detection technology of sedative residues in aquatic products[J]. Chinese Agricultural Science Bulletin, 2021, 37(12):86-91.
[12] 张璇,杨光昕,孔聪,等.高效液相色谱-串联质谱法测定水产品中镇静剂及其代谢物残留[J].分析化学研究报告,2021,49(3):460-467.
[12] ZHANG X, YANG GX, KONG C, et al. Determination of tranquillizer and their metabolites residues in aquatic products by high performance liquid chromatography-tandem mass spectrometry[J]. Chinese Journal of Analytical Chemistry, 2021, 49(3):460-467.
[13] 于慧娟, 钱蓓蕾, 黄冬梅, 等. 液相色谱串联质谱法测定大菱鱼和鳜鱼体中地西泮及其代谢物残留的研究[J]. 中国渔业质量与标准, 2011, 1(1):54-59.
[13] YU HJ, QIAN BL, HUANG DM, et al. Determination of diazepam and its residue in turbot and mandarinfish by high performance liquid chromatography-tandem mass spectrometry[J]. Chinese Fishery Quality and Standards, 2011, 1(1):54-59.
[14] 钱晓东, 于慧娟, 惠芸华, 等. 水产品中镇静剂残留的高效液相色谱-串联质谱法测定[J]. 湖南农业科学, 2010, (19):134-137.
[14] QIAN XD, YU HJ, HUI YH, et al. Determination of five kinds of sedative residues in aquatic products by high performance liquid chromatography-tandem mass spectrometry[J]. Hunan Agricultural Sciences, 2010, (19):134-137.
[15] 何晓明, 余鹏飞, 刘强欣, 等. 改良QuEChERS-高效液相色谱-串联质谱法同时测定水产品中的13种镇静剂[J]. 食品工业科技, 2020, 41(24):203-209.
[15] HE XM, YU PF, LiU QX, et al. Simultaneous determination of 13 sedative residues in aquatic products by modified QuEChERS combined with high-performance liquid chromatography-tandem mass spectrometry[J]. Science and Technology of Food Industry, 2020, 41(24):203-209.