 刘柏林,谢继安,赵紫微,王秀莉,单晓梅.超高压液相色谱-电喷雾串联四极杆质谱内标法同时测定禽类食品中11种喹诺酮类药物[J].中国食品卫生杂志,2017,29(3):316-321.
刘柏林,谢继安,赵紫微,王秀莉,单晓梅.超高压液相色谱-电喷雾串联四极杆质谱内标法同时测定禽类食品中11种喹诺酮类药物[J].中国食品卫生杂志,2017,29(3):316-321.DOi:10.13590/j.cjfh.2017.03.013
超高压液相色谱-电喷雾串联四极杆质谱内标法同时测定禽类食品中11种喹诺酮类药物
(安徽省疾病预防控制中心,安徽 合肥230601)
作者简介: 刘柏林男主管技师研究方向为食品安全的理化检验 E-mail:liubolin087@163.com
收稿日期: 2017-04-19
摘要:目的 建立同时测定禽类食品中11种喹诺酮类药物残留量的超高压液相色谱-电喷雾串联四极杆质谱(UPLC-ESI-MS/MS)分析方法。方法鸡肉和鸡蛋样品经1%乙酸乙腈(1∶100,V/V)混合液提取,冷冻离心与正己烷脱脂净化,以10 mmol/L乙酸铵(含0.1%甲酸)-甲醇为流动相,采用电喷雾电离,正离子扫描,多离子反应监测(MRM)模式,基质匹配同位素内标,标准曲线法定量。结果11种喹诺酮类药物在0.05~50.0 μg/L的浓度范围内线性良好,相关系数R2≥0.997 4,检出限范围为0.05~0.10 μg/kg,平均回收率为82.4%~104.3%,相对标准偏差小于15.0%。结论该方法的样品前处理过程简单,净化效果好,减少了基质效应的影响,适用于大批量禽类食品中喹诺酮类药物残留量的快速测定。
关键词:
超高压液相色谱-电喷雾串联四极杆质谱; 喹诺酮; 鸡蛋; 鸡肉; 同位素内标; 兽药残留; 食品污染物; 检测
中图分类号: R155 文献标识码:A 文章编号:1004-8456(2017)03-0316-06
Simultaneous determination of eleven quinolones drug residues in poultry products
by ultra performance liquid chromatography-electrospray ionization tandem
quadrupole mass spectrometry with isotope-labelled internal standards
by ultra performance liquid chromatography-electrospray ionization tandem
quadrupole mass spectrometry with isotope-labelled internal standards
(Anhui Provincial Center for Disease Control and Prevention,Anhui Hefei 230601,China)
Abstract:Objective A method for the determination of eleven quinolones drug residues in chicken and egg by the ultra performance liquid chromatography-electrospray ionization tandem quadrupole mass spectrometry (UPLC-ESI-MS/MS) was established. MethodsThe analytes in chicken and egg were extracted by 1% acetic acid-acetonitrile, and the fat were separated by centrifugation and extraction using hexane. The eleven analytes were separated by the mobile phase of 10 mmol/L ammonium acetate (contain 0.1% formic acid)-methanol on gradient elution. The identifications were achieved by electrospray ionization in positive mode (ESI +) using multiple reaction monitoring (MRM). The quantifications were performed by the matrix matched isotope-labelled internal standards. ResultsThe calibration curves of the eleven analytes showed a good linearity in the range of 0.05-50.0 μg/L with R2 above 0.997 4. Recoveries were between 82.4% and 104.3% with RSD less than 15.0%. The limits of detection were 0.05-0.10 μg/kg. ConclusionThe method had the advantages of simple pretreatment and low matrix effects, which was suitable for the rapid, high-throughput quantitative analysis of quinolones drug residues in poultry food.
Key words:
Ultra performance liquid chromatography-electrospray ionization tandem quadrupole mass spectrometry; quinolones; egg; chicken; isotope-labelled internal standards; veterinary drug residues; food contaminants; detection
喹诺酮类(quinolones,QNs)药物是一类人工合成的抗菌药,具有抗菌谱广、高效、低毒、组织穿透力强、价格低廉等优点[1],广泛用于禽兽及水产养殖疾病的治疗和预防。过量使用会导致药物在动物体内蓄积,现已报道动物源性食品中已有检出[2-3],对人类的健康构成了危害,残留问题已引起人们的广泛关注。联合国粮农组织/世界卫生组织食品添加剂专家联合委员会、欧盟已制定了多种喹诺酮类药物在动物组织中的最高残留限量[4],美国食品和药物管理局规定了家禽肉中的部分喹诺酮类药物限量,我国农业部公告第235号[5]对达氟沙星、二氟沙星、恩诺沙星、氟甲喹、恶喹酸、沙拉沙星制定了最高残留限量。
检测喹诺酮类残留的常见方法有高效液相色谱(HPLC)法[6-7]、高效液相色谱-串联质谱(HPLC-MS/MS)法[8-11]。HPLC-MS/MS具有选择性强、灵敏度高等特点,成为喹诺酮类残留分析的首选方法,当前现行检测喹诺酮类残留的国家标准方法为液相色谱-串联质谱(LC-MS/MS)法[12-14]。但多数方法采用外标法定量,内标法定量报道比较少[15-18]。外标法定量存在很强的基质干扰问题。本试验在前人研究的基础上,建立了超高压液相色谱-串联质谱(UPLC-MS/MS)法同时检测禽类食品中11种喹诺酮类药物残留的方法,该方法采用同位素内标法定量,且每个化合物均有对应的同位素内标,解决了几个化合物共用同一同位素内标带来的校正偏差的问题,有效校正了基质效应所引起的定量误差,降低了样品在提取、净化等过程中待测物的损失,大大提高了分析效率,保证了检测结果的准确性,适合大批量样品中喹诺酮类药物残留的快速测定。
11种喹诺酮类标准物质:氧氟沙星(CAS:82419-36-1)、培氟沙星(CAS:70458-92-3)、诺氟沙星(CAS:70458-96-7)、环丙沙星(CAS:85721-33-1)、洛美沙星(CAS:98079-51-7)、达氟沙星(CAS:119478-55-6)、双氟沙星(CAS:91296-86-5)、恩诺沙星(CAS:93106-60-6)、氟甲喹(CAS:42835-25-6)、恶喹酸(CAS:14698-29-4)、沙拉沙星(CAS:98105-99-8)均购自德国Dr.Ehrenstorfer;11种同位素内标准物质:氧氟沙星-D3(CAS:1173147-91-5)、培氟沙星-D5(CAS:1228182-51-1)、诺氟沙星-D5(CAS:1015856-57-1)、环丙沙星-D8(CAS:1130050-35-9)、洛美沙星-D5(CH021-25)、达氟沙星-D3(CAS:1217683-55-0)、双氟沙星-D3(CAS:1173021-89-0)、恶喹酸-D5(CAS:1189890-98-9)均购自百灵威科技,恩诺沙星-D5(CAS:1173021-92-5)、沙拉沙星-D8(CAS:1352879-52-7)均购自德国Dr. Ehrenstorfer,氟甲喹-13C3(CAS:1185049-09-5,美国TRC);甲醇、甲酸、乙腈、氨水、乙酸铵均为色谱纯。
混合标准工作液:分别准确移取上述标准储备液10 μl移至10 ml容量瓶中,用5%甲醇水溶液(含0.1%甲酸)稀释至刻度,混匀,配制成100 ng/ml的混合标准工作液;分别准确移取上述同位素内标标准储备液10 μl移至10 ml容量瓶中,用5%甲醇水溶液(含0.1%甲酸)稀释至刻度,混匀,配制成100 ng/ml 的混合内标工作液,现配现用。
标准系列应用液:分别准确移取上述混合标准工作溶液(100 ng/ml)5、10、50、100、200、500、1 000、5 000 μl移至10 ml容量瓶中,加入500 μl混合内标工作液,用5%甲醇水溶液(含0.1%甲酸)定容至刻度,配制成0.05、0.1、0.5、1.0、2.0、5.0、10.0、50.0 μg/L的标准系列应用液。
准确称取2.0 g(精确到0.01 g)均质样品置于50 ml具塞离心管中,加入50 μl同位素混合内标工作液,涡旋混匀,再加入10 ml 1%乙酸乙腈(1∶100,V/V),涡旋1 min,超声波提取10 min,4 ℃ 10 000 r/min离心5 min,取上清液于另一个离心管中。残留物再重复提取一次,合并两次提取液,加入5.0 g无水硫酸钠,4 ℃ 10 000 r/min离心5 min 后取上清液于40 ℃下氮气吹干。准确加入1.0 ml 5%甲醇水溶液(含0.1%甲酸),涡旋振荡溶解残留物,冷冻离心后(4 ℃ 10 000 r/min离心5 min),取上层液,再加入2.0 ml正己烷,涡旋混合30 s,转移至15 ml具塞离心管中,5 000 r/min离心5 min,弃去上层,下层液体经0.45 μm滤膜过滤,供UPLC-MS/MS测定。
质谱:离子源为电喷雾离子源(ESI),电离模式为ESI+,检测方式为多反应监测(MRM),毛细管电压3.8 kV,离子源温度150 ℃,脱溶剂气温度
动物源性食品中脂肪含量高,往往会影响回收率和测定结果的稳定性,用乙腈提取时,许多干扰物也会被提取出来,油脂是主要干扰物,为了去除基质中的脂肪类干扰物,采用高速冷冻离心除脂法,在低温状态下脂肪粒以固体状态从溶液中析出,以达到净化分离效果。复溶后,加入正己烷液液分配除去不饱和脂肪酸类油脂杂质,冷冻离心与正己烷脱脂两个净化步骤相结合,能有效地去除样品中的油脂类干扰物。
添加0.05 μg/kg浓度时,各待测物峰的信噪比(S/N)均大于3,故将其定为检出限;定量限定义为在获得理想的回收率和标准偏差条件下,可以检测到的样品溶液中目标化合物的最低浓度[9],本方法的定量限为0.5 μg/kg,该浓度下的添加回收率与相对标准偏差(RSD)均能够满足GB/T 27404—2008《实验室质量控制规范 食品理化检测》[22]的相关要求。
我国农业部2002年235号公告[5]中限量标准最严的是沙拉沙星,其限值为10.0 μg/kg。按此限量标准进行加标试验,考察该方法是否满足各化合物标准限值,称取2.0 g(精确到0.01 g)阴性样品,添加混合标准工作液,使加标水平分别达到1.0、5.0和10.0 μg/kg,加入混合内标工作液,按照1.2.2样品前处理方法进行处理,上机测定,每个添加水平进行3次平行测定。结果如表4所示,回收率范围为82.4%~104.3%,RSD范围为1.0%~14.6%。
检测喹诺酮类残留的常见方法有高效液相色谱(HPLC)法[6-7]、高效液相色谱-串联质谱(HPLC-MS/MS)法[8-11]。HPLC-MS/MS具有选择性强、灵敏度高等特点,成为喹诺酮类残留分析的首选方法,当前现行检测喹诺酮类残留的国家标准方法为液相色谱-串联质谱(LC-MS/MS)法[12-14]。但多数方法采用外标法定量,内标法定量报道比较少[15-18]。外标法定量存在很强的基质干扰问题。本试验在前人研究的基础上,建立了超高压液相色谱-串联质谱(UPLC-MS/MS)法同时检测禽类食品中11种喹诺酮类药物残留的方法,该方法采用同位素内标法定量,且每个化合物均有对应的同位素内标,解决了几个化合物共用同一同位素内标带来的校正偏差的问题,有效校正了基质效应所引起的定量误差,降低了样品在提取、净化等过程中待测物的损失,大大提高了分析效率,保证了检测结果的准确性,适合大批量样品中喹诺酮类药物残留的快速测定。
1材料与方法
1.1主要仪器与试剂
ACQUITYTM UPLC超高压液相色谱仪、ZEVO TQ MS串联质谱仪、8010G样品均质杯均购自美国Waters,Milli-Q超纯水系统,漩涡混匀器。11种喹诺酮类标准物质:氧氟沙星(CAS:82419-36-1)、培氟沙星(CAS:70458-92-3)、诺氟沙星(CAS:70458-96-7)、环丙沙星(CAS:85721-33-1)、洛美沙星(CAS:98079-51-7)、达氟沙星(CAS:119478-55-6)、双氟沙星(CAS:91296-86-5)、恩诺沙星(CAS:93106-60-6)、氟甲喹(CAS:42835-25-6)、恶喹酸(CAS:14698-29-4)、沙拉沙星(CAS:98105-99-8)均购自德国Dr.Ehrenstorfer;11种同位素内标准物质:氧氟沙星-D3(CAS:1173147-91-5)、培氟沙星-D5(CAS:1228182-51-1)、诺氟沙星-D5(CAS:1015856-57-1)、环丙沙星-D8(CAS:1130050-35-9)、洛美沙星-D5(CH021-25)、达氟沙星-D3(CAS:1217683-55-0)、双氟沙星-D3(CAS:1173021-89-0)、恶喹酸-D5(CAS:1189890-98-9)均购自百灵威科技,恩诺沙星-D5(CAS:1173021-92-5)、沙拉沙星-D8(CAS:1352879-52-7)均购自德国Dr. Ehrenstorfer,氟甲喹-13C3(CAS:1185049-09-5,美国TRC);甲醇、甲酸、乙腈、氨水、乙酸铵均为色谱纯。
1.2方法
1.2.1标准溶液的制备
标准储备液:分别称取适量的标准物质及同位素内标物质,用甲醇溶解,并定容至刻度,配制成100 μg/ml的标准储备液,避光保存于-20 ℃冰箱。混合标准工作液:分别准确移取上述标准储备液10 μl移至10 ml容量瓶中,用5%甲醇水溶液(含0.1%甲酸)稀释至刻度,混匀,配制成100 ng/ml的混合标准工作液;分别准确移取上述同位素内标标准储备液10 μl移至10 ml容量瓶中,用5%甲醇水溶液(含0.1%甲酸)稀释至刻度,混匀,配制成100 ng/ml 的混合内标工作液,现配现用。
标准系列应用液:分别准确移取上述混合标准工作溶液(100 ng/ml)5、10、50、100、200、500、1 000、5 000 μl移至10 ml容量瓶中,加入500 μl混合内标工作液,用5%甲醇水溶液(含0.1%甲酸)定容至刻度,配制成0.05、0.1、0.5、1.0、2.0、5.0、10.0、50.0 μg/L的标准系列应用液。
1.2.2样品前处理
鸡肉样品经高速组织捣碎机均匀绞碎,用四分法缩分出适量样品,均分成两份,转入清洁容器内,加封后作出标记,一份用于分析,另一份用于留样,于-20 ℃条件下保存。鸡蛋样品去壳混合均匀,转入清洁容器内,加封后作出标记,一份用于分析,另一份用于留样,于-20 ℃条件下保存。准确称取2.0 g(精确到0.01 g)均质样品置于50 ml具塞离心管中,加入50 μl同位素混合内标工作液,涡旋混匀,再加入10 ml 1%乙酸乙腈(1∶100,V/V),涡旋1 min,超声波提取10 min,4 ℃ 10 000 r/min离心5 min,取上清液于另一个离心管中。残留物再重复提取一次,合并两次提取液,加入5.0 g无水硫酸钠,4 ℃ 10 000 r/min离心5 min 后取上清液于40 ℃下氮气吹干。准确加入1.0 ml 5%甲醇水溶液(含0.1%甲酸),涡旋振荡溶解残留物,冷冻离心后(4 ℃ 10 000 r/min离心5 min),取上层液,再加入2.0 ml正己烷,涡旋混合30 s,转移至15 ml具塞离心管中,5 000 r/min离心5 min,弃去上层,下层液体经0.45 μm滤膜过滤,供UPLC-MS/MS测定。
1.2.3仪器条件
UPLC:色谱柱为Waters AcquityTM UPLC BEH C18柱(2.1 mm×100 mm,1.7 μm),柱温40 ℃,样品室温度10 ℃,进样体积10 μl,流动相A为10 mmol/L乙酸铵(含0.1%甲酸)、流动相B为甲醇,流速0.3 ml/min,梯度洗脱条件见表1。质谱:离子源为电喷雾离子源(ESI),电离模式为ESI+,检测方式为多反应监测(MRM),毛细管电压3.8 kV,离子源温度150 ℃,脱溶剂气温度
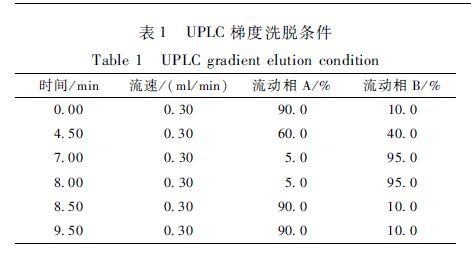
|
表1UPLC梯度洗脱条件 Table 1UPLC gradient elution condition |
1.2.4加标回收率试验
分别称取已绞碎均匀的阴性样品2.0 g(精确到0.01g)于3个50 ml离心管中,分别按1.0、5.0和10.0 μg/kg浓度添加11种喹诺酮混合标准工作液,提取步骤同1.2.2样品前处理。
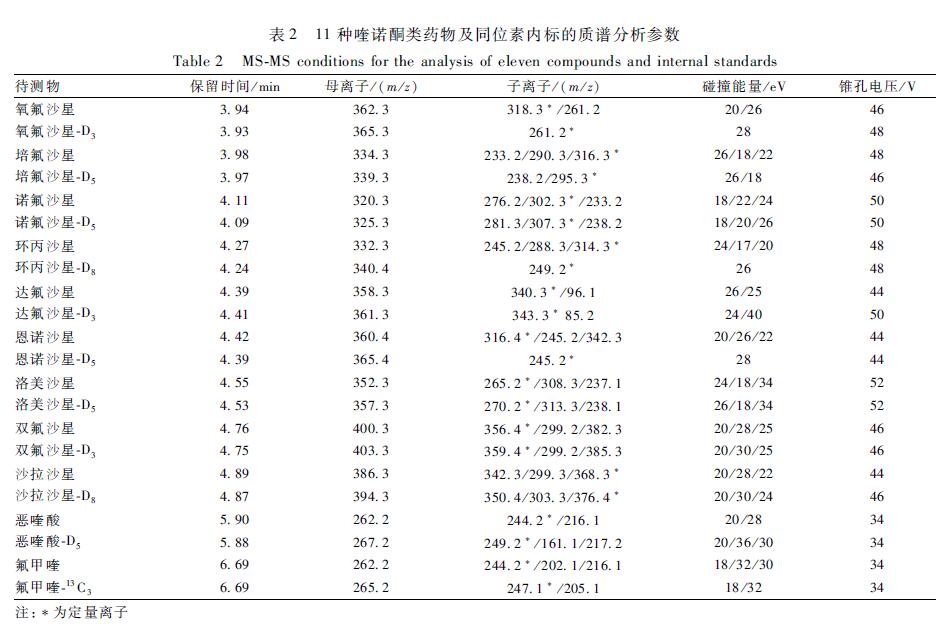
|
表211种喹诺酮类药物及同位素内标的质谱分析参数 Table 2MS-MS conditions for the analysis of eleven compounds and internal standards |
2结果与分析
2.1流动相的选择
本试验比较了0.1%甲酸水-乙腈、5 mmol/L乙酸铵(含0.1%甲酸)-乙腈、0.1%甲酸水-甲醇、0.1%甲酸水-甲醇乙腈溶液(40∶60,V/V)、5 mmol/L乙酸铵(含0.1%甲酸)-甲醇、10 mmol/L乙酸铵(含0.1%甲酸)-甲醇等6个流动相体系对分离的影响。据文献[8]报道,流动相的pH值对喹诺酮类药物的分离和保留具有明显影响,使用纯水为流动相时,11种喹诺酮类药物不能完全分离,其中氟甲喹和恶喹酸几乎是同一时间出峰,加入少量的甲酸控制流动相的pH值,不仅能提高待测物分离度,还能提供质子,提高正离子的灵敏度,试验比较了不同浓度甲酸对灵敏度的影响,甲酸含量为0.1%时,11种喹诺酮类药物的峰强度最大,增加甲酸含量,峰强度几乎不变,故选用0.1%甲酸水。水相中加入电解质乙酸铵,在保持较好分离度的前提下改善了离子峰的峰形,可以获得对称性好的色谱峰;另外,正离子模式下,与乙腈相比,甲醇能提供质子,增加离子强度,提高灵敏度;因此采用10 mmol/L乙酸铵(含0.1%甲酸)-甲醇作为流动相,使用表1的梯度洗脱,实现了11种喹诺酮的完全分离,且各峰形尖锐、对称性好(见图1)。
2.2样品前处理的优化
GB/T 20366—2006《动物源产品中喹诺酮类残留量的测定 液相色谱-串联质谱》[12]采用2%甲酸乙腈提取,乙腈饱和,正己烷脱脂,过微孔滤膜进样;GB/T 21312—2007《动物源性食品中14种喹诺酮药物残留检测方法 液相色谱-质谱/质谱法》[13]采用0.1 mol/L EDTA-Mcllvaine(EDTA为乙二胺四乙酸二钠,Mcllvaine为0.1 mol/L柠檬酸溶液与0.2 mol/L磷酸氢二钠溶液的混合缓冲液)缓冲溶液提取,HLB柱净化。本试验在溶液、乙腈与0.1mol/L EDTA-Mcllvaine缓冲液4种提取溶剂的提取效果。结果显示,采用0.1 mol/L EDTA-Mcllvaine缓冲溶液提取样品时,提取液浑浊且黏稠,杂质多,易堵塞HLB柱,向缓冲溶液中加入乙腈,沉淀蛋白,提取液变澄清,随乙腈含量的增加,沉淀蛋白效果明显,但使用HLB柱净化时,提取液需要稀释以降低乙腈含量,过程繁琐。另外,喹诺酮类药物是两性化合物,在纯乙腈中加入乙酸有利于待测物溶解在提取液中,提高回收率,本试验优化了乙酸的含量,发现使用1%的乙酸时,待测物的加标回收率最高;因此,采用1%乙酸乙腈(1∶100,V/V)进行提取。

|
图111种喹诺酮标准物质及同位素内标的色谱图(浓度为5.0 ng/ml) Figure 1Chromatograms of eleven quinolones drug residues and isotope-labelled internal standards |
2.3基质效应的消除
在应用UPLC-MS/MS测定基质复杂样品时,基质通常对分析物的离子化具有增强或抑制效应,可能严重影响定量的准确性[19]。本试验考察了基质效应对喹诺酮类药物的影响,以阴性样品提取液作为溶剂,配制5.0 ng/ml的混合标准工作液,测定其峰面积为A;以初始流动相为溶剂,配制5.0 ng/ml的混合标准工作液测定其峰面积为B。基质效应ME(%)=B/A×100%[20],其中ME>100%为基质增强效应,ME<100%为基质抑制效应[21]。表3可以看出,样品提取液对目标化合物存在一定的基质影响。本方法选取11种待测物同位素内标校正,按照1.2.2样品前处理的方法进行处理,制定标准曲线,由于样品溶液有已知浓度的内标,从而减少了样品基质效应的影响,确保方法的灵敏度、重现性和定量分析准确性。
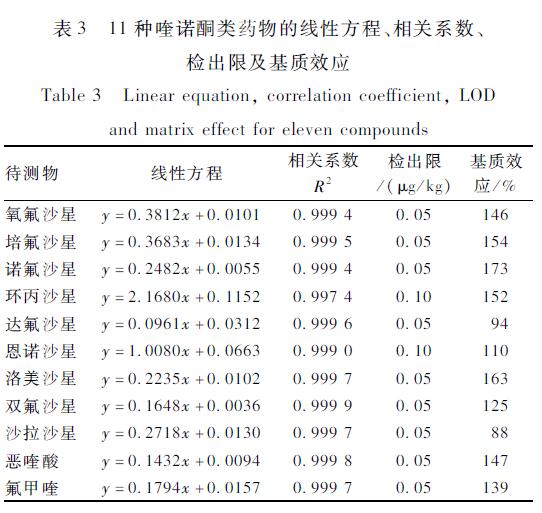
|
表311种喹诺酮类药物的线性方程、相关系数、 检出限及基质效应 Table 3Linear equation, correlation coefficient, LOD and matrix effect for eleven compounds |
2.4线性关系、检出限、精密度和回收率
选取阴性空白样品,加入标准系列应用液,按照1.2.2样品前处理的步骤进行处理,上机测定,以待测物峰面积(y)为纵坐标,质量浓度(x,μg/L)为横坐标,绘制标准曲线,得到线性回归方程(见表3),各待测物的浓度在0.05~50.0 μg/L的范围内与其峰面积呈良好的线性关系,相关系数R2均≥0.997 4。添加0.05 μg/kg浓度时,各待测物峰的信噪比(S/N)均大于3,故将其定为检出限;定量限定义为在获得理想的回收率和标准偏差条件下,可以检测到的样品溶液中目标化合物的最低浓度[9],本方法的定量限为0.5 μg/kg,该浓度下的添加回收率与相对标准偏差(RSD)均能够满足GB/T 27404—2008《实验室质量控制规范 食品理化检测》[22]的相关要求。
我国农业部2002年235号公告[5]中限量标准最严的是沙拉沙星,其限值为10.0 μg/kg。按此限量标准进行加标试验,考察该方法是否满足各化合物标准限值,称取2.0 g(精确到0.01 g)阴性样品,添加混合标准工作液,使加标水平分别达到1.0、5.0和10.0 μg/kg,加入混合内标工作液,按照1.2.2样品前处理方法进行处理,上机测定,每个添加水平进行3次平行测定。结果如表4所示,回收率范围为82.4%~104.3%,RSD范围为1.0%~14.6%。

|
表4样品中11种喹诺酮类残留的加标回收率及精密度(n=3) Table 4Recoveries and the relative standard deviation of eleven analytes in the actual samples |
2.5实际样品的测定
应用本方法分别测定鸡肉与鸡蛋样品,结果发现,样品中检出恩诺沙星、氧氟沙星、环丙沙星和诺氟沙星,其余各组分均未检出,实际检出率低于10%[鸡肉为6.8%(6/88),鸡蛋为8.8%(7/80)],含量为1.75~42.9 μg/kg,均未超出农业部2002年235号公告[5]中限量标准。
3小结
本试验建立了禽类食品中11种喹诺酮类残留检测的UPLC-MS/MS分析方法。通过对UPLC条件和样品前处理条件的优化,采用1%乙酸乙腈(1∶100,V/V)提取样品,冷冻离心与正己烷脱脂两步净化和同位素内标法补偿基质效应,使得方法的灵敏度、准确度和精密度均能满足兽药残留分析要求。本方法样品前处理简单,回收率高,适合批量样品的快速测定。
参考文献
[1]岳振峰,林秀云,唐少冰,等. 高效液相色谱-串联质谱法测定动物组织中的16种喹诺酮类药物残留[J]. 色谱, 2007,25(4):491-495.
[2]魏博娟,钱卓真,吴成业. 高效液相色谱-串联质谱法测定水产品中喹诺酮类药物残留[J]. 中国食品卫生杂志, 2011,23(3):249-254.
[3]包晓丽,任一平,张虹. 超高效液相色谱-电喷雾串联四极杆质谱法检测牛奶中22种喹诺酮类抗菌素[J]. 分析化学, 2009,37(3):389-394.
[4]HERNANDEZ-ARTESEROS J A, BARBOSA J, COMPANO R, et al. Analysis of quinolone residues in edible animal products[J]. Journal of Chromatography A, 2002,945(1/2):1-24.
[5]中华人民共和国农业部.中华人民共和国农业部公告:第235号[A/OL].(2002-12-24)[2017-02-20]. http://www.moa.gov.cn/zwllm/tzgg/gg/200302/t20030226_59300.htm.
[6]RODRGUEZ-DAZ R C, FERNNDEZ-ROMERO J M, AGUILAR-CABALLOS M P, et al. Determination of fluoroquino-lones in milk samples by post column derivatization liquid chroma-tography with luminescence detection[J]. J Agric Food Chem, 2006,54(26):9670-9676.
[7]中华人民共和国农业部,中华人民共和国国家卫生和计划生育委员会. 食品安全国家标准 牛奶中喹诺酮类药物多残留的测定 高效液相色谱法:GB 29692—2013[S].北京:中国标准出版社,2013.
[8]王志杰,冷凯良,孙伟红,等. 高效液相色谱-串联质谱法同时测定鳗鱼和虾中残留的33种喹诺酮和磺胺类药物[J]. 色谱, 2009,27(2):138-143.
[9]宋伟,胡艳云,韩芳,等. 超高效液相色谱-串联质谱法同时测定鸡肉中的二氯二甲吡啶酚、磺胺类和喹诺酮类药物残留[J]. 色谱, 2013,31(12):1161-1166.
[10]鞠玲燕,宋晓华,谷婕,等.超滤管净化/高效液相色谱-串联质谱法测定动物源性食品中喹诺酮类药物残留[J].分析测试学报,2016,35(1):42-47.
[11]马建民,夏曦,李晓薇,等. 阴离子交换固相萃取-超高效液相色谱-串联质谱法检测猪肌肉中13种喹诺酮类药物[J]. 中国食品卫生杂志, 2013,25(3):249-253.
[12]中华人民共和国国家质量监督检验检疫总局,中国国家标准化管理委员会. 动物源产品中喹诺酮类残留量的测定 液相色谱-串联质谱:GB/T 20366—2006[S].北京:中国标准出版社,2006.
[13]中华人民共和国国家质量监督检验检疫总局,中国国家标准化管理委员会. 动物源性食品中14种喹诺酮药物残留检测方法 液相色谱-质谱/质谱法:GB/T 21312—2007[S].北京:中国标准出版社,2007.
[14]中华人民共和国国家质量监督检验检疫总局,中国国家标准化管理委员会. 蜂蜜中19种喹诺酮类药物残留量的测定方法 液相色谱-质谱/质谱法:GB/T 23412—2009 [S].北京:中国标准出版社,2009.
[15]李丽莉,罗轶,何颂华,等. 高效液相色谱-串联质谱法测定鱼肉中8种喹诺酮类药物的残留量[J]. 中国食品卫生杂志, 2012,24(1):37-40.
[16]王志杰,冷凯良,孙伟红,等. 高效液相色谱-串联质谱内标法同时测定水产品中15种喹酮类药物残留量[J]. 分析科学学报, 2010,26(4):409-414.
[17]祝子铜,徐佳文,雷美康,等. HPLC-MS-MS同位素内标法测定火腿中14种喹诺酮类残留量[J]. 食品科学, 2014,35(20):258-264.
[18]张颖颖,李莹莹. 超高效液相色谱-串联质谱测定猪肉中16种喹诺酮药物残留量[J]. 肉类研究, 2016,30(5):36-41.
[19]李锋格,苏敏,李晓岩,等. 分散固相萃取-超高效液相色谱-串联质谱法测定鸡肝中磺胺类、喹诺酮类和苯并咪唑类药物及其代谢物的残留量[J]. 色谱, 2011,29(2):120-125.
[20]MATUSZEWSKI B K, CONSTANZER M L, CHAVEZ-ENG C M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS[J]. Anal Chem, 2003,75(13):3019-3030.
[21]何建丽,彭涛,谢洁,等. 高效液相色谱-串联质谱法测定动物肝脏中20种全氟烷基类化合物[J]. 分析化学, 2015,43(1):40-48.
[22]中华人民共和国国家质量监督检验检疫总局,中国国家标准化管理委员会. 实验室质量控制规范 食品理化检测:GB/T 27404—2008[S].北京:中国标准出版社,2008.
[2]魏博娟,钱卓真,吴成业. 高效液相色谱-串联质谱法测定水产品中喹诺酮类药物残留[J]. 中国食品卫生杂志, 2011,23(3):249-254.
[3]包晓丽,任一平,张虹. 超高效液相色谱-电喷雾串联四极杆质谱法检测牛奶中22种喹诺酮类抗菌素[J]. 分析化学, 2009,37(3):389-394.
[4]HERNANDEZ-ARTESEROS J A, BARBOSA J, COMPANO R, et al. Analysis of quinolone residues in edible animal products[J]. Journal of Chromatography A, 2002,945(1/2):1-24.
[5]中华人民共和国农业部.中华人民共和国农业部公告:第235号[A/OL].(2002-12-24)[2017-02-20]. http://www.moa.gov.cn/zwllm/tzgg/gg/200302/t20030226_59300.htm.
[6]RODRGUEZ-DAZ R C, FERNNDEZ-ROMERO J M, AGUILAR-CABALLOS M P, et al. Determination of fluoroquino-lones in milk samples by post column derivatization liquid chroma-tography with luminescence detection[J]. J Agric Food Chem, 2006,54(26):9670-9676.
[7]中华人民共和国农业部,中华人民共和国国家卫生和计划生育委员会. 食品安全国家标准 牛奶中喹诺酮类药物多残留的测定 高效液相色谱法:GB 29692—2013[S].北京:中国标准出版社,2013.
[8]王志杰,冷凯良,孙伟红,等. 高效液相色谱-串联质谱法同时测定鳗鱼和虾中残留的33种喹诺酮和磺胺类药物[J]. 色谱, 2009,27(2):138-143.
[9]宋伟,胡艳云,韩芳,等. 超高效液相色谱-串联质谱法同时测定鸡肉中的二氯二甲吡啶酚、磺胺类和喹诺酮类药物残留[J]. 色谱, 2013,31(12):1161-1166.
[10]鞠玲燕,宋晓华,谷婕,等.超滤管净化/高效液相色谱-串联质谱法测定动物源性食品中喹诺酮类药物残留[J].分析测试学报,2016,35(1):42-47.
[11]马建民,夏曦,李晓薇,等. 阴离子交换固相萃取-超高效液相色谱-串联质谱法检测猪肌肉中13种喹诺酮类药物[J]. 中国食品卫生杂志, 2013,25(3):249-253.
[12]中华人民共和国国家质量监督检验检疫总局,中国国家标准化管理委员会. 动物源产品中喹诺酮类残留量的测定 液相色谱-串联质谱:GB/T 20366—2006[S].北京:中国标准出版社,2006.
[13]中华人民共和国国家质量监督检验检疫总局,中国国家标准化管理委员会. 动物源性食品中14种喹诺酮药物残留检测方法 液相色谱-质谱/质谱法:GB/T 21312—2007[S].北京:中国标准出版社,2007.
[14]中华人民共和国国家质量监督检验检疫总局,中国国家标准化管理委员会. 蜂蜜中19种喹诺酮类药物残留量的测定方法 液相色谱-质谱/质谱法:GB/T 23412—2009 [S].北京:中国标准出版社,2009.
[15]李丽莉,罗轶,何颂华,等. 高效液相色谱-串联质谱法测定鱼肉中8种喹诺酮类药物的残留量[J]. 中国食品卫生杂志, 2012,24(1):37-40.
[16]王志杰,冷凯良,孙伟红,等. 高效液相色谱-串联质谱内标法同时测定水产品中15种喹酮类药物残留量[J]. 分析科学学报, 2010,26(4):409-414.
[17]祝子铜,徐佳文,雷美康,等. HPLC-MS-MS同位素内标法测定火腿中14种喹诺酮类残留量[J]. 食品科学, 2014,35(20):258-264.
[18]张颖颖,李莹莹. 超高效液相色谱-串联质谱测定猪肉中16种喹诺酮药物残留量[J]. 肉类研究, 2016,30(5):36-41.
[19]李锋格,苏敏,李晓岩,等. 分散固相萃取-超高效液相色谱-串联质谱法测定鸡肝中磺胺类、喹诺酮类和苯并咪唑类药物及其代谢物的残留量[J]. 色谱, 2011,29(2):120-125.
[20]MATUSZEWSKI B K, CONSTANZER M L, CHAVEZ-ENG C M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS[J]. Anal Chem, 2003,75(13):3019-3030.
[21]何建丽,彭涛,谢洁,等. 高效液相色谱-串联质谱法测定动物肝脏中20种全氟烷基类化合物[J]. 分析化学, 2015,43(1):40-48.
[22]中华人民共和国国家质量监督检验检疫总局,中国国家标准化管理委员会. 实验室质量控制规范 食品理化检测:GB/T 27404—2008[S].北京:中国标准出版社,2008.