 韩小敏,徐文静,赵熙,张宏元,张靖,李凤琴.玉米及其制品和小麦及其制品中白僵菌素和恩镰孢菌素的高效液相色谱-串联质谱检测方法的建立[J].中国食品卫生杂志,2017,29(6):633-640.
韩小敏,徐文静,赵熙,张宏元,张靖,李凤琴.玉米及其制品和小麦及其制品中白僵菌素和恩镰孢菌素的高效液相色谱-串联质谱检测方法的建立[J].中国食品卫生杂志,2017,29(6):633-640. DOi:10.13590/j.cjfh.2017.06.001
玉米及其制品和小麦及其制品中白僵菌素和恩镰孢菌素的高效液相色谱-串联质谱检测方法的建立
(国家食品安全风险评估中心 卫生部食品安全风险评估重点实验室,北京100021)
收稿日期:2017-11-24
作者简介:韩小敏女副研究员研究方向为食品卫生E-mail:hanxiaomin@cfsa.net.cn
通信作者:李凤琴女研究员研究方向为食品卫生E-mail:lifengqin@cfsa.net.cn
基金项目:科技基础工作专项(2013FY113400);北京市自然科学基金(7163235)
摘要:目的建立了玉米及其制品和小麦及其制品中白僵菌素(beauvericin,BEA)、恩镰孢菌素A(enniatin A,ENA)、恩镰孢菌素A1(enniatin A1,ENA1)、恩镰孢菌素B(enniatin B,ENB)和恩镰孢菌素B1(enniatin B1,ENB1)的高效液相色谱-串联质谱检测方法。方法样品经乙腈-水(85∶15,V/V)提取、室温静置、固相萃取柱净化后,以2 mmol/L乙酸铵水-乙腈作为流动相,采用电喷雾电离正离子多反应监测模式进行检测。结果该方法对玉米及其制品和小麦及其制品中BEA和4种恩镰孢菌素(ENNs)的检出限范围分别为0.01~0.12和0.02~0.21 μg/kg,定量限范围分别为0.02~0.44和0.05~0.68 μg/kg,平均加标回收率范围分别为91.6%~149.7%和100.5%~128.2%,基质抑制或增强效应分别为88.0%~101.5%和55.8%~106.5%,且BEA和4种ENNs在所测基质中线性关系和精密度良好,相关系数均>0.99,相对标准偏差均<15%。结论所建立的方法操作简便、灵敏度高、准确性好,可用于玉米及其制品和小麦及其制品中BEA和4种ENNs的测定。
关键词:
玉米及其制品; 小麦及其制品; 高效液相色谱-串联质谱; 白僵菌素; 恩镰孢菌素; 真菌毒素; 食品污染物
文章编号:1004-8456(2017)06-0633-08
中图分类号:R155
文献标志码:A
Development of high performance liquid chromatography tandem-mass spectrometry method for determination of beauvericin and enniatins in corn and wheat and their products
(Key Laboratory of Food Safety Risk Assessment of Ministry of Health,China National Center for Food Safety Risk Assessment,Beijing 100021,China)
Abstract:ObjectiveA high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) for determination of beauvercin (BEA), enniatin A (ENA), enniatin A1 (ENA1), enniatin B (ENB) and enniatin B1 (ENB1) in corn, wheat and their products was developed. MethodsSamples were extracted with acetonitrile-water (85∶ 15, V/V), cleaned up with solid phase extraction column followed by HPLC-MS/MS determination with electrospray positive ionization (ESI+) under multiple reaction monitoring (MRM) mode, and with 2 mmol/L ammonium acetate water-acetonitrile as the mobile phase. ResultsThe limits of detection for BEA and 4 kinds of enniatins (ENNs) ranged from 0.01 to 0.12 μg/kg for corn and corn products and 0.02 to 0.21 μg/kg for wheat and wheat products, and the limits of quantification ranged from 0.02 to 0.44 μg/kg and 0.05 to 0.68 μg/kg for both cereals. The mean recoveries ranged from 91.6% to 149.7% for corn and corn products and 100.5% to 128.2% for wheat and wheat products, respectively. The matrix induced suppression or enhancement effect ranged from 88.0% to 101.5% and 55.8% to 106.5% for both cereals, respectively. The method for BEA and ENNs determination showed good linearity and precision in all matrixes with correlation coefficient above 0.99, and the relative standard deviations were less than 15%. ConclusionThe method developed was of simple operation, high sensitivity and good accuracy, which could be used for BEA and 4 kinds of ENNs determination in corn and wheat and their products.
Key words:
Corn and their products; wheat and their products; high performance liquid chromatography-tandem mass spectrometry; beauvercin; enniatins; mycotoxin; food contamination
白僵菌素(beauvericin,BEA)和恩镰孢菌素(enniatins,ENNs)主要是由镰刀菌属的燕麦镰刀菌(F.avenaceum)和木贼镰刀菌(F.equiseti)等的某些菌种侵染小麦、大麦、黑麦和燕麦等谷物后,在潮湿和低温条件下产生的真菌毒素[1-2]。到目前为止,已经发现了29种ENNs,但食品中最常见的ENNs主要有4种即恩镰孢菌素A(enniatin A,ENA)、恩镰孢菌素A1(enniatin A1,ENA1)、恩镰孢菌素B(enniatin B,ENB)和恩镰孢菌素B1(enniatin B1,ENB1)[3-4]。据报道[5-7],BEA和ENNs具有基因毒性和细胞毒性,可诱导染色体畸变、姊妹染色单体交换和微核形成等。目前,已在西班牙、突尼斯、意大利、日本、伊朗、巴西、摩洛哥、挪威和丹麦等国家的谷物及其制品中检测到这两类毒素[8-14]。UHLIG等[13]对2000—2002年挪威产73份燕麦、75份大麦和80份小麦共计228份样品中BEA和4种ENNs(ENA、ENA1、ENB和ENB1)的污染调查发现,228份样品中5种毒素均有检出。SRENSEN等[14]对丹麦2005—2006年收获的80份玉米和玉米青贮饲料中BEA和4种ENNs(ENA、ENA1、ENB和ENB1)的污染调查表明,两类样品中4种ENNs的检出率由高到低依次为ENB>ENB1>ENA1>ENA。考虑到BEA和ENNs污染的严重性和普遍性,近年来人们对其关注也越来越多。
基于液液萃取或固相萃取的高效液相色谱(HPLC)配合光电二极管阵列检测器(DAD)的HPLC-DAD法是检测食品中BEA和ENNs常用方法[15]。但由于单纯的色谱分析方法存在特异性不强、灵敏度不高等特点,其应用越来越受到限制。近年来,基于HPLC与质谱联用技术开发的高效液相色谱-串联质谱(HPLC-MS/MS)因结合了色谱优越的分离能力与质谱优越的定性能力,明显地提高了检测的灵敏度和特异性,具有检出限低、能获取待测化合物的分子结构信息、对前处理要求不高等特点,成为目前分析食品中BEA和ENNs的最佳手段[16-19]。但我国目前尚未建立食品中两类毒素的检测方法,本课题组前期在对目前国际上食品中BEA和ENNs的污染与分析方法的最新研究进行概述的基础上[19],提出了采用HPLC-MS/MS同时测定谷物及其制品中BEA和4种ENNs(ENA、ENA1、ENB和ENB1)的方法。
BEA(BIA-B1238)、ENA(BIA-E1165)、ENA1(BIA-E1166)、ENB(BIA-E1167)和ENB1(BIA-E1168)均为固体粉末标准品(纯度均≥97%),均购自澳大利亚Bioaustralis;甲醇、乙腈、醋酸、乙酸铵均为质谱纯,超纯水由本实验室的超纯水仪制备。
混合标准储备液:从ENA、ENA1、ENB、ENB1和BEA标准储备液中分别移取300、100、100、100 和100 μl 至10 ml容量瓶,乙腈定容至刻度,混匀。制备成ENA为150 μg/L、ENA1和ENB均为400 μg/L、ENB1和BEA均为1 000 μg/L的混合标准储备液,4 ℃下避光保存。
混合标准工作液:从ENA、ENA1、ENB、ENB1和BEA标准储备液中分别移取30、10、10、10和10 μl 至10 ml容量瓶,乙腈定容至刻度,混匀,制备成ENA为15 μg/L、ENA1和ENB均为40 μg/L、ENB1和BEA均为100 μg/L的混合标准工作液。4 ℃下避光保存,用于标准系列工作溶液的配制。
标准系列工作溶液:移取一定量混合标准工作液用乙腈按比例稀释,配制成一系列标准工作液,其中ENA浓度为0.005、0.015、0.05、0.15、0.5、1.5、5、15 μg/L;ENA1和ENB浓度均为0.013、0.04、0.13、0.4、1.33、4.0、13.33和40 μg/L;ENB1和BEA的浓度均为0.033、0.1、0.33、1.0、3.33、10.0、33.33和100 μg/L。若配制基质匹配标准工作溶液,则将稀释溶剂由乙腈更换为空白基质提取液,各毒素浓度保持不变。
提取:称取5.0 g样品(精确至0.01 g)于50 ml刻度离心管中,加入40 ml样品提取液(乙腈∶水=85∶15,V/V),加盖密封后涡旋混匀30~60 s,180 r/min振荡提取30 min后,室温静置15~20 min;准确移取10 ml上清液于另一50 ml刻度离心管,加入20 ml水进行稀释,加盖密封并涡旋混匀30~60 s,室温静置15~20 min备用。
净化:取4 ml 最终稀释后的样品提取液全部通过事先依次用3 ml甲醇和3 ml水预先活化的SPE柱,依次用3 ml 10%乙腈水溶液、3 ml 50%乙腈水溶液淋洗并抽干SPE柱。最后用2 ml 90%乙腈水溶液洗脱毒素,抽干SPE柱并收集洗脱液,涡旋混匀后备用。
色谱:色谱柱:Waters ACQUITY UPLC BEH C18柱(2.1 mm×50 mm,1.7 μm),柱温35 ℃,样品室温度15 ℃,进样体积5 μl,流速0.2 ml/min。流动相A为含2 mmol/L乙酸铵的水溶液,流动相B为乙腈。梯度洗脱程序:0~2 min 100%A,2~3 min 100%A~40%A,3~19 min 40%A~30%A,19~21 min 30%A~100%A,20~20.1 min 100%A。
ESI+条件下,分别考察甲醇-水-醋酸(10∶89∶1,V/V)、2 mmol/L乙酸铵水溶液作为流动相A和甲醇-水-醋酸(97∶2∶1,V/V)、乙腈作为流动相B对BEA和4种ENNs保留时间和质谱信号响应值的影响。

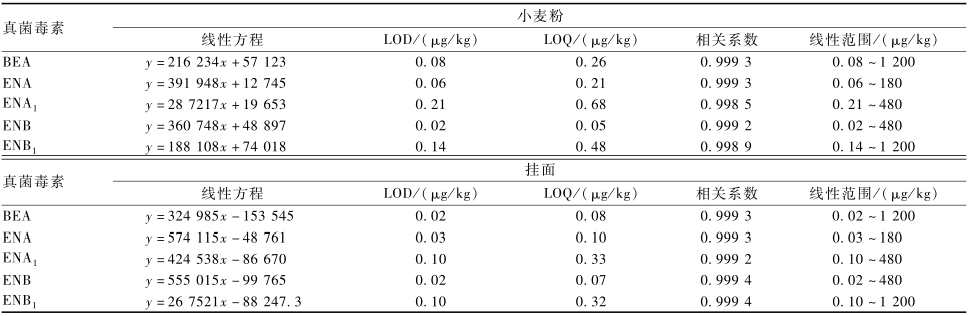
性范围分别为0.03~1 200、0.01~180、0.09~480、0.01~480和0.08~1 200 μg/kg,玉米渣中BEA、 ENA、ENA1、ENB和ENB1的线性范围分别为0.03~400、0.02~60、0.09~160、0.01~160和0.07~ 400 μg/kg。小麦及其制品中BEA、ENA、ENA1、ENB和ENB1的线性范围分别为0.02~1 200、0.03~180、0.10~480、0.02~480和0.10~1 200 μg/kg,玉米及其制品和小麦及其制品中5种真菌毒素的LOD范围分别为0.01~0.12和0.02~0.21 μg/kg,LOQ范围分别为0.02~0.44和0.05~0.68 μg/kg,且BEA和4种ENNs在不同基质中的线性关系良好,线性相关系数均>0.99,可满足玉米及其制品和小麦及其制品中5种真菌毒素定量检测的要求。
4种ENNs进行提取和净化,同一天不同时间点连续进样5次,同时对这些样品连续测定5 d,分别计算各毒素色谱峰的峰面积的RSD,进行日间和日内精 密度的考察,结果见表8。可以看出,玉米及其制品的日内和日间RSD分别为0.2%~6.9%和1.9%~13.5%,小麦及其制品的日内和日间RSD分别
为0.1%~6.8%和3.9%~13.3%,表明所建方法具有良好的精密度。
基于液液萃取或固相萃取的高效液相色谱(HPLC)配合光电二极管阵列检测器(DAD)的HPLC-DAD法是检测食品中BEA和ENNs常用方法[15]。但由于单纯的色谱分析方法存在特异性不强、灵敏度不高等特点,其应用越来越受到限制。近年来,基于HPLC与质谱联用技术开发的高效液相色谱-串联质谱(HPLC-MS/MS)因结合了色谱优越的分离能力与质谱优越的定性能力,明显地提高了检测的灵敏度和特异性,具有检出限低、能获取待测化合物的分子结构信息、对前处理要求不高等特点,成为目前分析食品中BEA和ENNs的最佳手段[16-19]。但我国目前尚未建立食品中两类毒素的检测方法,本课题组前期在对目前国际上食品中BEA和ENNs的污染与分析方法的最新研究进行概述的基础上[19],提出了采用HPLC-MS/MS同时测定谷物及其制品中BEA和4种ENNs(ENA、ENA1、ENB和ENB1)的方法。
1材料与方法
1.1材料
1.1.1样品来源
玉米及其制品包括玉米粒、玉米渣和玉米面,小麦及其制品包括小麦粉和挂面,均购自部分省(市)超市及农贸市场。
1.1.2主要仪器与试剂
QTRAPTM5500 HPLC-MS/MS(配备有ExionLC系统和QTRAPTM5500质谱仪,美国AB Sciex)、Sep-Pak Vac C18固相萃取柱(SPE,200 mg,3 ml,美国Waters)、涡旋混合器、圆周式振荡器、低温离心机。BEA(BIA-B1238)、ENA(BIA-E1165)、ENA1(BIA-E1166)、ENB(BIA-E1167)和ENB1(BIA-E1168)均为固体粉末标准品(纯度均≥97%),均购自澳大利亚Bioaustralis;甲醇、乙腈、醋酸、乙酸铵均为质谱纯,超纯水由本实验室的超纯水仪制备。
1.2方法
1.2.1BEA和ENNs标准溶液的配制
标准储备液:分别称取一定量的BEA、ENA、ENA1、ENB和ENB1固体粉末标准品,用乙腈溶解并定容后混匀,得到ENA标准储备液(5 μg/ml)、ENA1标准储备液(40 μg/ml)、ENB标准储备液(40 μg/ml)、ENB1标准储备液(100 μg/ml)和BEA标准储备液(100 μg/ml),-20 ℃下避光保存,备用。混合标准储备液:从ENA、ENA1、ENB、ENB1和BEA标准储备液中分别移取300、100、100、100 和100 μl 至10 ml容量瓶,乙腈定容至刻度,混匀。制备成ENA为150 μg/L、ENA1和ENB均为400 μg/L、ENB1和BEA均为1 000 μg/L的混合标准储备液,4 ℃下避光保存。
混合标准工作液:从ENA、ENA1、ENB、ENB1和BEA标准储备液中分别移取30、10、10、10和10 μl 至10 ml容量瓶,乙腈定容至刻度,混匀,制备成ENA为15 μg/L、ENA1和ENB均为40 μg/L、ENB1和BEA均为100 μg/L的混合标准工作液。4 ℃下避光保存,用于标准系列工作溶液的配制。
标准系列工作溶液:移取一定量混合标准工作液用乙腈按比例稀释,配制成一系列标准工作液,其中ENA浓度为0.005、0.015、0.05、0.15、0.5、1.5、5、15 μg/L;ENA1和ENB浓度均为0.013、0.04、0.13、0.4、1.33、4.0、13.33和40 μg/L;ENB1和BEA的浓度均为0.033、0.1、0.33、1.0、3.33、10.0、33.33和100 μg/L。若配制基质匹配标准工作溶液,则将稀释溶剂由乙腈更换为空白基质提取液,各毒素浓度保持不变。
1.2.2样品前处理
样品粉碎:(1)散装样品:将玉米及其制品和小麦及其制品用四分法缩分至500 g,如有必要则先用高速粉碎机粉碎,-20 ℃下避光保存备用。(2)定型包装样品:独立包装≤500 g的样品,如有必要粉碎则直接用高速粉碎机粉碎后作为样品,备用;若单个样品的独立包装>500 g,则先用四分法缩分至500 g,如有必要粉碎则再用高速粉碎机粉碎后作为样品,备用,-20 ℃下避光保存。提取:称取5.0 g样品(精确至0.01 g)于50 ml刻度离心管中,加入40 ml样品提取液(乙腈∶水=85∶15,V/V),加盖密封后涡旋混匀30~60 s,180 r/min振荡提取30 min后,室温静置15~20 min;准确移取10 ml上清液于另一50 ml刻度离心管,加入20 ml水进行稀释,加盖密封并涡旋混匀30~60 s,室温静置15~20 min备用。
净化:取4 ml 最终稀释后的样品提取液全部通过事先依次用3 ml甲醇和3 ml水预先活化的SPE柱,依次用3 ml 10%乙腈水溶液、3 ml 50%乙腈水溶液淋洗并抽干SPE柱。最后用2 ml 90%乙腈水溶液洗脱毒素,抽干SPE柱并收集洗脱液,涡旋混匀后备用。
1.2.3仪器条件
质谱:选择电喷雾正离子扫描(ESI+)、多反应监测(MRM)模式,驻留时间为200 ms,离子源参数见表1,BEA和4种ENNs的保留时间及MRM参数见表2。

|
表1BEA和ENNs测定的离子源参数 Table 1Ion source parameters for BEA and ENNs determination 注:气帘气、雾化气、辅助加热气分别为30、80、80 psi |
|
|
表2HPLC-MS/MS测定BEA和ENNs的保留时间和MRM参数 Table 2Retention time and MRM parameters for BEA and ENNs determination by HPLC-MS/MS 注:*为定量子离子;DP为去簇电压;CE为碰撞能量;CXP为碰撞室射出电压;EP为射入电压 |
ESI+条件下,分别考察甲醇-水-醋酸(10∶89∶1,V/V)、2 mmol/L乙酸铵水溶液作为流动相A和甲醇-水-醋酸(97∶2∶1,V/V)、乙腈作为流动相B对BEA和4种ENNs保留时间和质谱信号响应值的影响。
1.3数据处理
采用MultiQuantTM3.0.2软件对每份样品中各毒素色谱峰的峰面积、线性关系、校正曲线、信噪比(S/N)、检出限(LOD)和定量限(LOQ)等进行计算。后续的数据处理包括平均加标回收率、基质抑制或增强效应(基质效应,SSE)、精密度等采用Microsoft Excel 2007进行计算。不同基质中每种毒素的平均加标回收率(RA)和基质效应分别按照公式(1)和(2)进行计算,LOD和LOQ分别按3倍S/N和10倍S/N进行计算。
2结果与分析
2.1质谱条件的优化
BEA和4种ENNs均为环脂肽类真菌毒素,本试验分别考察了ESI+和ESI-条件下BEA和4种ENNs的[M+H]+、[M+Na]+和[M+NH4]+母离子峰的信号响应值强弱,根据欧盟2002/657/EC指令[20]对于低分辨率质谱联用检测的规定每种毒素选择两个适宜的子离子,后以三通连接方式将标准品与流动相同时进样,优化离子源参数。经过优化发现,5种毒素在ESI+条件下母离子峰的信号响应值均高于在ESI-条件下母离子峰的信号响应值,ESI+条件下5种毒素均以[M+NH4]+峰的信号响应值最优,优化后的BEA、ENA、ENA1、ENB和ENB1的母离子峰分别为801.4、699.5、685.5、657.5、671.5,其他参数具体见表1和2。
2.2色谱条件的优化
液相色谱流动相的组成不仅影响BEA和4种ENNs的色谱行为,还会影响BEA和4种ENNs的离子化效率。根据2.1质谱条件优化的结果,ESI+条件下分别考察了甲醇-水-醋酸(10∶89∶1,V/V)和甲醇-水-醋酸(97∶2∶1,V/V)作为流动相与2 mmol/L乙酸铵水溶液和乙腈作为流动相时对5种真菌毒素质谱信号响应值的影响。因5种毒素[M+NH4]+峰的信号响应值明显高于[M+H]+和[M+Na]+峰的信号响应值,所以2 mmol/L乙酸铵水溶液和乙腈作为流动相时5种毒素的信号相应值明显高于甲醇-水-醋酸(10∶89∶1,V/V)和甲醇-水-醋酸(97∶2∶1,V/V)作为流动相时的信号相应值,且2 mmol/L乙酸铵水溶液和乙腈作为流动相时各毒素色谱峰的峰形更对称,基线信号强度更低(见图1)。因乙酸铵浓度过高会影响色谱柱寿命,所以后续试验中选择2 mmol/L乙酸铵水溶液作为流动相A、乙腈作为流动相B进行洗脱。综合考虑各毒素的分离度、保留时间等方面的影响,确定本试验流动相的流速和梯度洗脱程序。
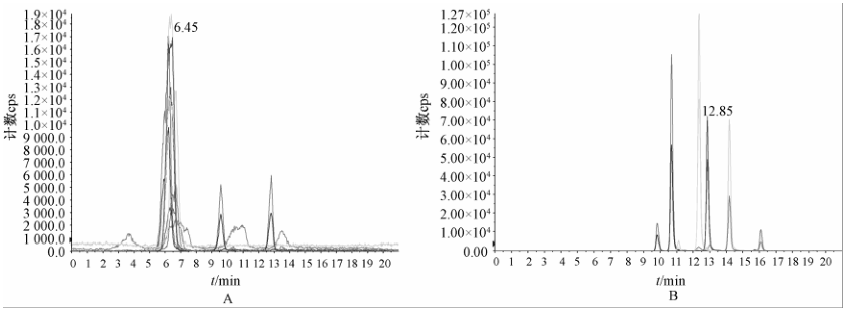
|
注:A表示流动相为甲醇-水-醋酸(10∶89∶1,V/V)和甲醇-水-醋酸(97∶2∶1,V/V);B表示流动相为2 mmol/L乙酸铵水溶液和乙腈 图1ESI+条件下BEA和ENNs在不同流动相中的总离子流图 Figure 1TIC of BEA and ENNs obtained by different mobile phases under ESI+ |
2.3样品前处理条件的优化
本试验在SPE柱常规操作步骤和已有文献[9]报道的基础上,重点考察了玉米粒和挂面中两类毒素的提取效率。采用两种方法(1)提取后静置15~20 min和(2)1 400×g低温离心10 min对平均加标回收率的影响。结果发现,振荡提取30 min后温室静置15~20 min与1 400×g低温离心10 min对玉 米粒和挂面样品中毒素的平均加标回收率均差异无统计学意义(P>0.05),具体结果见表3。考虑到大量样品中BEA和4种ENNs的提取时,低温离心会明显延长操作时间并增加操作步骤的复杂性,因此后续试验中选择振荡提取30 min后温室静置15~20 min的方式进行BEA和4种ENNs的提取。
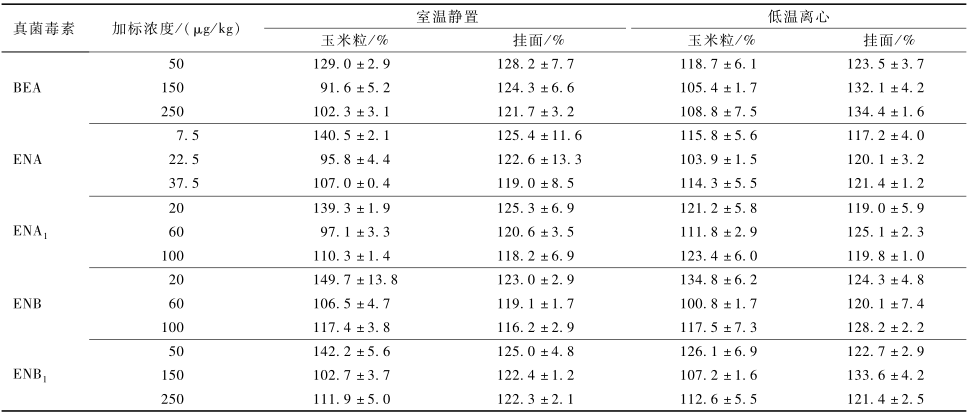
|
表3BEA和ENNs在不同前处理方式中的平均加标回收率(±s) Table 3Effect of different sample preparation on recoveries of BEA and ENNs |
2.4基质效应
基质效应是目前采用电喷雾电离分析毒素面临的一个巨大挑战。减少进入质谱系统基质组 分的含量和用合适的方法比如基质匹配标准曲线、标准品添加和同位素内标法对测定结果进行校正是目前常用的校正基质效应的方法。前者因在减少进入质谱系统基质组分含量的同时也降低了目标分析物的含量,且明显提高了目标化合物的LOD和LOQ等越来越不被人们采用。基质匹配标准曲线校正法是目前常用的测定食品中毒素含量时校正基质效应的方法。本试验采用HPLC-MS/MS对玉米及其制品包括玉米粒、玉米渣、玉米面和小麦及其制品包括小麦粉和挂面中BEA和4种ENNs的含量测定时发现,玉米及其制品的基质效应为88.0%~101.5%,小麦及其制品的基质效应为55.8%~61.5%,其中小麦粉的基质抑制效应最明显,为55.8%~61.5%,见表4。
2.5方法验证
2.5.1方法的线性范围、LOD和LOQ
根据欧盟SANCO方法学验证标准[21]对所建立的方法进行验证,具体见表5和6。可以看出,玉米粒和玉米面的BEA、ENA、ENA1、ENB和ENB1的线
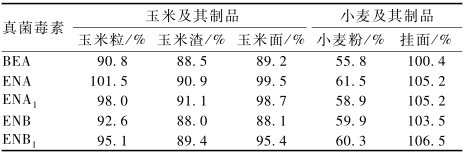
|
表4BEA和ENNs在玉米及其制品和小麦及其制品中的基质效应 Table 4Matrix effect on detection of BEA and ENNs in corn and wheat and their products |
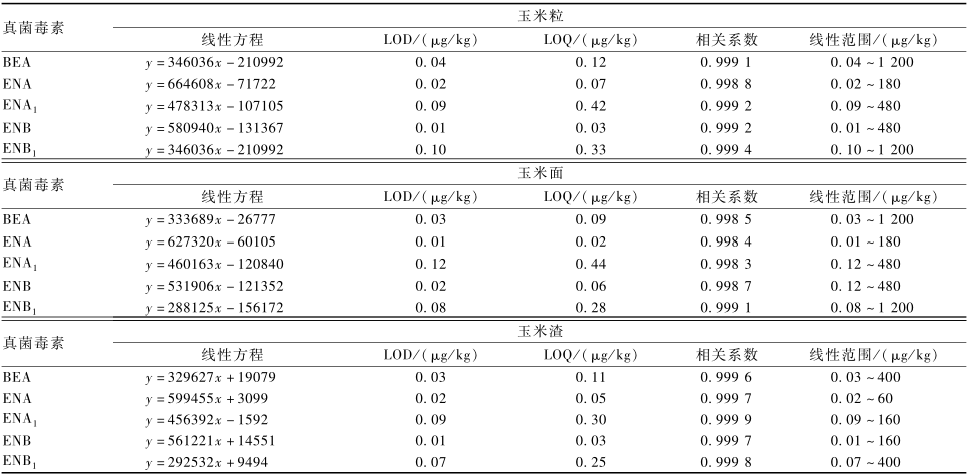
|
表5玉米及其制品中BEA和ENNs的相关技术指标 Table 5Related technical index of method for detection of BEA and ENNs in corn and corn products |
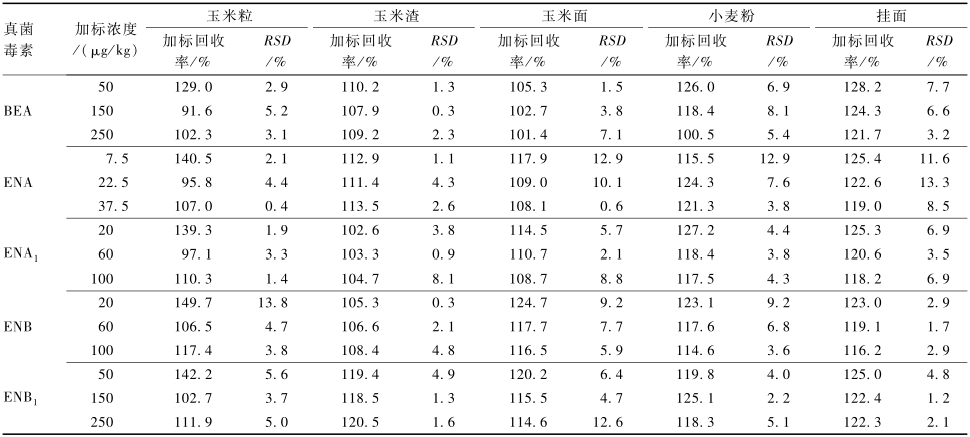
2.5.2方法的加标回收率
取不含BEA和ENNs的玉米及其制品和小麦及其制品,分别添加低、中、高3个浓度的BEA和ENNs混合标准溶液,每个加标水平进行3次重复试验,按1.2.2进行样品前处理,加标回收率结果见表7。可以看出,玉米及其制品和小麦及其制品中5种真菌毒素的平均加标回收率范围分别为91.6%~149.7%和100.5%~128.2%,相对标准偏差(RSD)范围分别为0.3%~13.8%和1.2%~13.3%,符合欧盟对食品中真菌毒素定量测定时对回收率及RSD的要求[22]。
2.5.3方法的精密度
本试验采用添加低、中和高3个浓度BEA和ENNs样品,按照1.2.2对加标样品中的BEA和
|
表6小麦及其制品中BEA和ENNs的相关技术指标 Table 6Related technical index of method for detection of BEA and ENNs in wheat and wheat products |
|
表7玉米及其制品和小麦及其制品中BEA和ENNs的加标回收率和RSD(n=3) Table 7Recoveries and relative standard deviation of method for detection of BEA and ENNs in corn and wheat and their products |

|
表8玉米及其制品和小麦及其制品中BEA和ENNs的日内和日间精密度 Table 8Intra-and inter-day precision of method for detection of BEA and ENNs in corn and wheat and their products |
2.6玉米及其制品和小麦及其制品中BEA和ENNs污染水平的测定
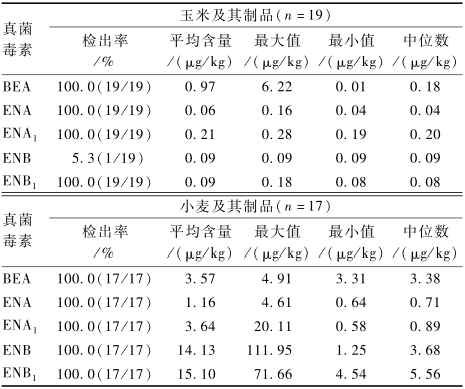
从北京、安徽、宁夏等省(市/自治区)采集市售玉米及其制品19份、小麦及其制品17份对所建立的方法进行验证,结果见表9。可以看出,所测的玉米及其制品中BEA和4种ENNs的污染水平均低于小麦及其制品中BEA和4种ENNs的污染水平。19份玉米及其制品中均检出BEA、ENA、ENA1和ENB1,平均含量和中位数分别为0.97和0.18、0.06和0.04、0.21和0.20、0.09和0.08 μg/kg,仅1份玉米样品中检出ENB,检出率为5.3%、含量为0.09 μg/kg。17份小麦及其制品中均检出BEA和4种ENNs,BEA、ENA、ENA1、ENB和ENB1的平均含量和中位数分别为3.57和3.38、1.16和0.71、3.64和0.89、14.13和3.68、15.10和5.56 μg/kg,值得注意的是一份来自宁夏的小麦粉样品中ENB和ENB1的含量高达111.95和71.66 μg/kg,但仍低于SRENSEN等[14]报道的丹麦2005—2006年产玉米和玉米青贮饲料中ENB和ENB1的最大污染水平,同时本次测定样品中玉米及其制品和小麦及其制品中BEA和4种ENNs的污染水平也均低于UHLIG等[13]报道的2000—2002年挪威产燕麦、大麦和小麦中BEA和4种ENNs的污染水平。
|
表9玉米及其制品和小麦及其制品中BEA和ENNs的污染状况 Table 9Natural occurrence of BEA and ENNs in corn and wheat samples |
3小结
本试验在优化液相色谱条件、质谱条件和样品前处理方法的基础上,对方法的准确性及精密度进行了验证,同时对实际样品中BEA和4种ENNs的含量进行测定,建立了玉米及其制品和小麦及其制品中BEA和4种ENNs含量测定的HPLC-MS/MS检测方法。与现有文献报道的谷物及其制品经乙腈-水-乙酸(79∶20∶1,V/V)一步法提取后直接经HPLC-MS/MS或HPLC检测BEA和4种ENNs的方法比较[10-11],该方法具有操作简便、灵敏度高、稳定性好、基质效应弱等特点,适于批量样品中BEA和4种ENNs的定性确证与定量检测,能够满足我国污染物监测中玉米及其制品和小麦及其制品中BEA和4种ENNs含量测定的要求。
参考文献
[1]LOGRIECO A,RIZZO A,FERRACANE R,et al. Occurrence of beauvericin and enniatins in wheat affected by Fusarium avenaceum head blight[J]. Appl Environ Microbiol,2002,68(1):82-85.
[2]JESTOI M. Emerging mycotoxins fusaproliferin,beauvericin,enniatins and moniliformin-a review[J]. Critical Reviews in Food Science and Nutrition,2008,48(1):21-49.
[3]HERRMANN M, ZOCHER R, HAESE A A. Enniatin production by Fusarium strains and its effect on potato tuber tissue[J]. Appl Environ Microbiol,1996,62(62):393-398.
[4]MALACHOVA A,DZUMAN Z,VEPRIKOVA Z,et al. Deoxynivalenol,deoxynivalenol-3-glucoside and enniatins:the major mycotoxins found in cereal-based products on the Czech market[J]. Journal of Agricultural and Food Chemistry,2011,59(24):12990-12997.
[5]CELIK M,AKSOY H,YILMAZ S. Evaluation of beauvericin genotoxicity with the chromosomal aberrations,sister-chromatid exchanges and micronucleus assays[J]. Ecotoxicology and Environmental Safety,2010,73(7):1553-1557.
[6]TAN D C,FLEMATTI G R,GHISALBERTI E L,et al. Toxigenicity of enniatins from western Australian Fusarium species to brine shrimp(artemia franciscana)[J]. Toxicon,2011,57(5):817-825.
[7]HAMILL R L,HIGGENS C E,BOAZ H E,et al. The structure of beauvericin, a new desipeptide antibiotic toxic to artemia salina[J]. Tetrahedron Letters,1969,10(49):4255-4258.
[8]SERRANO A B,FONT G,RUIZ M J,et al. Co-occurrence and risk assessment of mycotoxins in food and diet from Mediterranean area[J]. Food Chemistry,2012,135(2):423-429.
[9]YOSHINARI T,SUZUKI Y,SUGITAKONISHI Y,et al. Occurrence of beauvericin and enniatins in wheat flour and corn grits on the Japanese market and their co-contamination with type B trichothecene mycotoxins[J]. Food Additives and Contaminants Part A Chemistry Analysis Control Exposure and Risk Assessment,2016,33(10):620-626.
[10]NAZARI F,SULYOK M,KOBARFARD F,et al. Evaluation of emerging Fusarium mycotoxins beauvericin,enniatins,fusaproliferin and moniliformin in domestic rice in Iran[J]. Iranian Journal of Pharmaceutical Research,2015,14(2):505-512.
[11]SOUZA M D L M D,SULYOK M,FREITASSILVA O,et al. Cooccurrence of mycotoxins in maize and poultry feeds from Brazil by liquid chromatography/tandem mass spectrometry[J]. Scientific World Journal,2013,2013(1):77-87.
[12]BLESA J,MOLT J C,AKHDARI S E,et al. Simultaneous determination of Fusarium mycotoxins in wheat grain from Morocco by liquid chromatography coupled to triple quadrupole mass spectrometry[J]. Food Control,2014,46(4):1-5.
[13]UHLIG S,TORP M,HEIER B T. Beauvericin and enniatins A,A1,B and B1 in Norwegian grain:a survey[J]. Food Chemistry,2006,94(2):193-201.
[14]SRENSEN J L,NIELSEN K F,RASMUSSEN P H,et al. Development of a LC-MS/MS method for the analysis of enniatins and beauvericin in whole fresh and ensiled maize[J]. Journal of Agricultural and Food Chemistry,2008,56(21):10439-10443.
[15]HERRMANN M,ZOCHER R,HAESE A A. Enniatin production by Fusarium strains and its effect on potato tuber tissue[J]. Appl Environ Microbiol,1996,62(62):393-398.
[16]HABLER K,RYCHLIK M. Multi-mycotoxin stable isotope dilution LC-MS/MS method for Fusarium toxins in cereals[J]. Analytical and Bioanalytical Chemistry,2016,218(1):447-454.
[17]HU L,RYCHLIK M. Biosynthesis of 15N3-labeled enniatins and beauvericin and their application to stable isotope dilution assays[J]. Journal of Agricultural and Food Chemistry,2012,60(29):7129-7136.
[18]HU L,RYCHLIK M. Occurrence of enniatins and beauvericin in 60 Chinese medicinal herbs [J]. Food Additives and Contaminants Part A Chemistry Analysis Control Exposure and Risk Assessment,2014,31(7):1240-1245.
[19]韩小敏,李凤琴,徐文静. 食品中白僵菌素和恩镰孢菌素污染与分析方法研究进展[J].中国食品卫生杂志,2017,29(4):508-513.
[20]European Commission. Commission Decision 2002/657/EC of 12 August 2002[J]. Off J Eur Communities L,2002,221:8-36.
[21]PIHLSTRM T. Method validation and quality control procedures for pesticide residue analysis in food and feed. Document No. SANCO/12495/2011[A].2011.
[22]Commission Regulation(EC)No 401/2006. Laying down the methods of sampling and analysis for the official control of the levels of mycotoxins in foodstuffs [S]. Official Journal of the European Union,2006,L70:31-33.
[2]JESTOI M. Emerging mycotoxins fusaproliferin,beauvericin,enniatins and moniliformin-a review[J]. Critical Reviews in Food Science and Nutrition,2008,48(1):21-49.
[3]HERRMANN M, ZOCHER R, HAESE A A. Enniatin production by Fusarium strains and its effect on potato tuber tissue[J]. Appl Environ Microbiol,1996,62(62):393-398.
[4]MALACHOVA A,DZUMAN Z,VEPRIKOVA Z,et al. Deoxynivalenol,deoxynivalenol-3-glucoside and enniatins:the major mycotoxins found in cereal-based products on the Czech market[J]. Journal of Agricultural and Food Chemistry,2011,59(24):12990-12997.
[5]CELIK M,AKSOY H,YILMAZ S. Evaluation of beauvericin genotoxicity with the chromosomal aberrations,sister-chromatid exchanges and micronucleus assays[J]. Ecotoxicology and Environmental Safety,2010,73(7):1553-1557.
[6]TAN D C,FLEMATTI G R,GHISALBERTI E L,et al. Toxigenicity of enniatins from western Australian Fusarium species to brine shrimp(artemia franciscana)[J]. Toxicon,2011,57(5):817-825.
[7]HAMILL R L,HIGGENS C E,BOAZ H E,et al. The structure of beauvericin, a new desipeptide antibiotic toxic to artemia salina[J]. Tetrahedron Letters,1969,10(49):4255-4258.
[8]SERRANO A B,FONT G,RUIZ M J,et al. Co-occurrence and risk assessment of mycotoxins in food and diet from Mediterranean area[J]. Food Chemistry,2012,135(2):423-429.
[9]YOSHINARI T,SUZUKI Y,SUGITAKONISHI Y,et al. Occurrence of beauvericin and enniatins in wheat flour and corn grits on the Japanese market and their co-contamination with type B trichothecene mycotoxins[J]. Food Additives and Contaminants Part A Chemistry Analysis Control Exposure and Risk Assessment,2016,33(10):620-626.
[10]NAZARI F,SULYOK M,KOBARFARD F,et al. Evaluation of emerging Fusarium mycotoxins beauvericin,enniatins,fusaproliferin and moniliformin in domestic rice in Iran[J]. Iranian Journal of Pharmaceutical Research,2015,14(2):505-512.
[11]SOUZA M D L M D,SULYOK M,FREITASSILVA O,et al. Cooccurrence of mycotoxins in maize and poultry feeds from Brazil by liquid chromatography/tandem mass spectrometry[J]. Scientific World Journal,2013,2013(1):77-87.
[12]BLESA J,MOLT J C,AKHDARI S E,et al. Simultaneous determination of Fusarium mycotoxins in wheat grain from Morocco by liquid chromatography coupled to triple quadrupole mass spectrometry[J]. Food Control,2014,46(4):1-5.
[13]UHLIG S,TORP M,HEIER B T. Beauvericin and enniatins A,A1,B and B1 in Norwegian grain:a survey[J]. Food Chemistry,2006,94(2):193-201.
[14]SRENSEN J L,NIELSEN K F,RASMUSSEN P H,et al. Development of a LC-MS/MS method for the analysis of enniatins and beauvericin in whole fresh and ensiled maize[J]. Journal of Agricultural and Food Chemistry,2008,56(21):10439-10443.
[15]HERRMANN M,ZOCHER R,HAESE A A. Enniatin production by Fusarium strains and its effect on potato tuber tissue[J]. Appl Environ Microbiol,1996,62(62):393-398.
[16]HABLER K,RYCHLIK M. Multi-mycotoxin stable isotope dilution LC-MS/MS method for Fusarium toxins in cereals[J]. Analytical and Bioanalytical Chemistry,2016,218(1):447-454.
[17]HU L,RYCHLIK M. Biosynthesis of 15N3-labeled enniatins and beauvericin and their application to stable isotope dilution assays[J]. Journal of Agricultural and Food Chemistry,2012,60(29):7129-7136.
[18]HU L,RYCHLIK M. Occurrence of enniatins and beauvericin in 60 Chinese medicinal herbs [J]. Food Additives and Contaminants Part A Chemistry Analysis Control Exposure and Risk Assessment,2014,31(7):1240-1245.
[19]韩小敏,李凤琴,徐文静. 食品中白僵菌素和恩镰孢菌素污染与分析方法研究进展[J].中国食品卫生杂志,2017,29(4):508-513.
[20]European Commission. Commission Decision 2002/657/EC of 12 August 2002[J]. Off J Eur Communities L,2002,221:8-36.
[21]PIHLSTRM T. Method validation and quality control procedures for pesticide residue analysis in food and feed. Document No. SANCO/12495/2011[A].2011.
[22]Commission Regulation(EC)No 401/2006. Laying down the methods of sampling and analysis for the official control of the levels of mycotoxins in foodstuffs [S]. Official Journal of the European Union,2006,L70:31-33.